
ゲムシタビンHCV治療のための投薬計画
ゲムシタビン(または、明細書中に記載されているその塩、プロドラッグもしくは誘導体)を約50mg/m2/日の用量範囲で1〜7日間(例えば、1、2、3、4、5、6または7日間)投与した後治療を休止することを含むC型肝炎ウイルス感染を含めたフラビウイルス科ウイルス感染を治療するための投薬計画。ウイルス量を場合により経時的にモニターし、休止後ウイルスリバウンドをモニターする。有意なウイルス量が再び観察されない限り治療を再開せず、観察されたときには1〜7日間、より好ましくは1〜3日間の治療を繰り返す。この治療は患者の健康をモニターし、維持するために無期限に継続され得る。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ゲムシタビン、またはその医薬的に許容され得る塩、プロドラッグもしくは誘導体を用いるフラビウイルスまたはペスチウイルス、特にC型肝炎ウイルス(HCV)を治療するための方法及び投薬計画である。
【背景技術】
【0002】
ゲムザール(登録商標)(ゲムシタビンHCl)は白血病及び各種固形腫瘍(例えば、膵臓癌、非小細胞肺癌、卵巣癌、乳癌、中皮腫等)に対して抗腫瘍活性を有するピリミジン代謝拮抗物質である。ゲムシタビンは式β−D−2’,2’−ジフルオロシチジン(下記構造式参照)を有するヌクレオシド類縁体である。ゲムシタビンは元々抗ウイルス効果について研究されたが、その後抗悪性腫瘍剤として開発された(D.C.Delong,L.W.Hertel及びJ.Tang,「2’,2’−ジフルオロデオキシシチジンの抗ウイルス活性(Antiviral activity of 2',2' Difluorodeoxycytidine)」,American Society of Microbiology(1986))。ゲムシタビンは食品医薬品局(FDA)により次の適応症に認可されている:(1)手術不能な局所的に進行した(IIIA期またはIIIB期)または転移(IV期)非小細胞肺癌(NSCLC)を患っている患者に対する第一選択治療としてシスプラチンと併用して、(2)膵臓の局所的に進行した(切除不能なII期またはIII期)または転移(IV期)腺癌を患っている患者に対する第一選択治療として、及び(3)5−フルオロウラシル(5−FU)の治療を受けた患者における膵臓癌に対する第二選択治療として。
【0003】
【化4】

【0004】
ゲムシタビン(dFdC)はDNA合成(S期)を経ている細胞を主に標的とする細胞周期特異的物質である。ゲムシタビンは細胞内で律速酵素のデオキシシチジンキナーゼ(dCK)によりそのモノホスフェート形態(dFdCMP)に代謝される(V.Heinemannら,「2’,2’−ジフルオロデオキシシチジン及び1−β−D−アラビノフラノシルシトシンの細胞薬物動態及び毒性の比較(Comparison of the Cellular Pharmacokinetics and Toxicity of 2',2'-Difluorodeoxycytidine and 1-Beta-D-Arabinofuranosylcytosine)」,Cancer Res.,48(14):4024−31(1988))。その後他のヌクレオシドキナーゼによりリン酸化されると、活性な代謝物dFdCDPdF及びdFdCTPが形成される。ゲムシタビンの細胞毒性はジホスフェート及びトリホスフェート代謝物による作用の組合せに起因し、前記代謝物によりゲムシタビンの致死作用が高められる。これらの作用を図1に要約する。まず、dFdCDPはリボヌクレオチドレダクターゼを阻害し(経路1)、これによりDNA複製に必要な細胞デオキシヌクレオチド(例えば、デオキシシチジントリホスフェート,dCTP)の濃度が低下する(図1;ゲムシタビン及びDNA修復の自己増強作用)。リボヌクレオチドレダクターゼの阻害により細胞dCTP濃度が低くなると、ゲムシタビン誘導致死率にとって重要な事象であるDNAへのdFdCTPアナログの取り込みが助けられる(経路2)(P.Huang及びW.Plunkett,「フルダラビン及びゲムシタビン誘導アポトーシス:アナログのDNAへの取り込みは重要な事象である(Fludarabine- and Gemcitabine-Induced Apoptosis: Incorporation Of Analogs Into DNA Is A Critical Event)」,Cancer Chemother.Pharmacol.,36(3):181−8(1995);P.Huang及びW.Plunkett,「ゲムシタビンによるアポトーシスの誘導(Indudction Of Apoptosis By Gemcitabine)」,Semin.Oncol.,22(4 補遺11):19−25(1995))。高dCTPレベルは律速酵素dCKを阻害するので、細胞dCTPレベルが低下するとゲムシタビンリン酸化速度も増加する(経路3)。dCKに対する阻害作用と対照的に、dCTPはゲムシタビンヌクレオチドの細胞からの消失のための律速酵素であるdCMPデアミナーゼ活性のために必要なコファクターである(経路4)。細胞毒性代謝物dFdCTPは直接dCMPデアミナーゼを阻害する(経路5)(Y.Z.Xu及びW.Plunkett,「2’,2’−ジフルオロデオキシシチジンの完全ヒト白血病細胞作用におけるデオキシシチジレートデアミナーゼの調節(Modulation Of Deoxycytidylate Deaminase In Intact Human Leukemia Cells Action of 2',2'-difluorodeoxycytidine)」,Biochem.Pharmacol.,44(9):1819−27(1992))。最後に、高細胞内濃度でFdCTPはCTPシンターゼを阻害し(経路6)、よってCTPの合成が阻止され、その結果dCTPの合成も阻止される(V.Heinemannら,「ゲムシタビン:細胞内ヌクレオチド及びデオキシヌクレオチド代謝の調節剤(Gemcitabine: A Modulator of Intracellular Nucleotide And Deoxynucleotide Metabolism)」,Semin.Oncol.,22(4 補遺11):11−8(1995))。
【0005】
ゲムシタビンは、dCKによるdFdCMPからのモノホスフェートへのリン酸化に対する良好な基質であり、デオキシシチジン(チャイニーズハムスター卵巣(CHO)細胞由来の部分的に精製した酵素を用いて測定して、Km=1.6μmol/L)と類似の基質効率(Vmax/Km)で3.6μmol/LのKmを示す(V.Heinemannら,「2’,2’−ジフルオロデオキシシチジン及び1−β−D−アラビノフラノシルシトシンの細胞薬物動態及び毒性の比較(Comparison of the Cellular Pharmacokinetics and Toxicity of 2',2'-Difluorodeoxycytidine and 1-Beta-D-Arabinofuranosylcytosine)」,Cancer Res.,48(14):4024−31(1988))。ゲムシタビンのリン酸化はその生物学的活性にとって必須であり、dCKを欠く細胞はゲムシタビンにより影響されない。DNA、RNA及びタンパク質合成の放射活性前駆体を用いる研究で、ゲムシタビンの効果は主にDNAに向けられることが立証された(W.Plunkettら,「ゲムシタビン:前臨床薬理学及び作用機序(Gemcitabine: Preclinical Pharmacology And Mechanisms Of Action)」,Semin.Oncol.,23(5 補遺10):3−15(1996))。DNA合成のモデル系で、トリホスフェートdFdCTPがヒトDNAポリメラーゼα及びεにより成長DNAプライマー鎖に組み込まれ、各ポリメラーゼが正常ヌクレオチド(dCTP)よりも20倍優先的であることを示すことが確認された(P.Huangら,「DNA合成に対する2’,2’−ジフルオロデオキシシチジンの作用(Action of 2',2'-Difluorodeoxycytidine on DNA Synthesis)」,Cancer Res.,51(22):6110−7(1991))。ユニークなことに、ゲムシタビンの取り込みの後1つ以上のヌクレオチドを付加した後にDNAポリメラーゼが阻害される。終わりから2番目の位置に置くと、3’→5’プロフリーディングエキソヌクレアーゼによるdFdCMPによる切除はdCMPの切除よりも非常にゆっくり進行する。“仮面連鎖停止”として記載されているこの現象により、ゲムシタビンのDNA複製及び修復を阻害する能力が改良され、ゲムシタビンのDNA障害剤(例えば、シスプラチン)との相乗作用のメカニズムが与えられる。
【0006】
ゲムシタビンは、臨床使用中に脱アミノ化生成物の2’,2’−ジフルオロデオキシウリジン(dFdU)への生体内変化を介する迅速な代謝クリアランスに関与する酵素である細胞内シチジンデアミナーゼ(Km=96μM)に対する良好な基質である(D.Y.Bouffard,J.Laliberte及びR.L.Momparler,「2’,2’−ジフルオロデオキシシチジン(ゲムシタビン)と精製ヒトデオキシシチジンキナーゼ及びシチジンデアミナーゼに関するカイネティック研究(Kinetic Studies On 2',2'-Difluorodeoxycytidine (Gemcitabine) With Purified Human Deoxycytidine Kinase And Cytidine Deaminase)」,Biochem.Pharmacol.,45(9):1857−91(1993))。ゲムシタビンは血液、肝臓、腎臓及び他の組織中で急速に脱アミノ化される。ゲムシタビン処分が放射標識した薬物100mg/m2を1回30分間注入により投与した5人の被験者において評価された。ゲムシタビン(<10%)及び不活性代謝物dFdUは排泄用量の99%を占めた。代謝物dFdUは血漿中でも検出され、ゲムシタビン血漿タンパク質結合は無視できた。
【0007】
ゲムシタビンの薬物動態をいろいろな固形腫瘍を患っている353人の患者(2/3が男性)で研究した。薬物動態パラメータは、短い(<70分)及び長い(70〜285分)注入時間で週1回投与し、周期的に数週間休薬して異なる期間治療した患者からのデータを用いて調べた。投与されたゲムシタビン総用量は500〜3600mg/m2であった。ゲムシタビン薬物動態は直線的であり、2−区画モデルにより記載されている。消失は腎排泄に依存し、クリアランスは年齢及び性の影響を受けた。1回及び複数回投与研究の集団薬物動態分析から、ゲムシタビンの分布容量は注入時間及び性により大きく影響されたことが分かった。注入時間が短いときのゲムシタビン半減期は32〜92分であり、注入時間が長いときのゲムシタビン半減期は245〜638分であった。これらのデータは注入時間を長くすると分布容量が多くなることを反映した。分布容量は注入時間が短い(<70分)ときには50L/m2であり、ゲムシタビンが組織中に広く分布しないことを示している。反対に、注入時間が長いときの分布容量は370L/m2に増加し、これはゲムシタビンが組織区画内でゆっくり平衡化することを反映している。代謝物は週投1回の投与で蓄積されなかったが、その消失は腎排泄に依存し、dFdUレベルは腎不全により影響され得る。
【0008】
ゲムシタビン処分に対する有意な腎または肝不全の影響は今まで評価されていない。活性代謝物dFdCTPは末梢血単核細胞から抽出され得、単核細胞からのdFdCTPの終相半減期は1.7〜19.4時間である。
【0009】
重要なことに、最大耐量(MTD)は注入スケジュール及び注入頻度に大きく依存する(E.Bovenら,「実験的ヒト癌における抗腫瘍効果に対するゲムシタビンのスケジュール及び用量の影響(The Influence Of The Schedule And The Dose Of Gemcitabine On The Antitumour Efficacy In Experimental Human Cancer)」,Br.J.Cancer,68(1):52−6(1993))。60分を越える注入時間の延長及び週1回以上の投与回数はゲムシタビン関連毒性を高めることが判明した。典型的には、骨髄抑制は白血病、血小板減少症及び貧血を発現する用量限定的毒性である。血液学的毒性のために用量を頻繁に調節する必要があるので、患者は治療中骨髄抑制についてモニターされなければならない。ゲムシタビンに関連する他の毒性には、口内炎、悪心、嘔吐、発熱、発疹、軽い不規則性機能亢進(parasthesias)、軽い脱毛、インフルエンザ様症状(すなわち、発熱、悪寒、筋肉痛、咳及び頭痛)、呼吸困難、浮腫、軽度の蛋白尿及び血尿、1つまたは両方の血清トランスアミナーゼの一過性上昇及び下痢が含まれる。2つの臨床治験において、局所的に進行した膵臓癌または転移性膵臓癌患者でゲムシタビンの効果を評価した。第1の治験では、過去に化学療法を受けたことのない患者でゲムシタビンと5−FUを比較し、第2の治験では過去に5−FUまたは5−FU含有投薬を用いる治療を受けたことのある患者を評価した。いずれの治験でも、ゲムシタビンは1000mg/m2の用量で30分間注入により週1回連続7週間(または、投薬中止が必要となる毒性が生ずるまで)投与した後1週間治療を休止した。その後のサイクルは週1回連続3週間注入した後、1週間休止とした。この研究での主たる効果パラメーターは、鎮痛剤消費量、痛み強度、パフォーマンス状態及び体重変化に基づく改良により定義され且つ測定される臨床的に有利な応答に基づいていた。第1の治験では、局所的に進行した膵臓癌または転移性膵臓癌患者に対してゲムシタビン及び5−FUを多施設において予測的一重盲検でランダム比較した(H.A.Burrisら,「進行膵臓癌患者に対する第一選択治療としてのゲムシタビンを用いる生存率及び臨床効果の改良:無作為化治験(Improvements in Survival And Clinical Benefit With Gemcitabine As First-Line Therapy For Patients With Advanced Pancreas Cancer: A Randomized Trial)」,J.Clin.Oncol.,15(6):2403−13(1997))。各治療群で評価した63患者で、ゲムシタビンは5−FUと比較して臨床効果応答、生存率及び病気進行までの時間の点で統計的に有意な向上を示した。第2の治験では、過去に5−FUまたは5−FU含有投薬の治療を受けたことのある63患者に対してゲムシタビンを多施設においてオープンラベル試験した(M.L.Rothenbergら,「5−FU耐性膵臓癌患者におけるゲムシタビンの第II相治験(A Phase II Trial Of Gemcitabine In Patients With 5-FU-Refractory Pancreas Cancer)」,Ann.Oncol.,7(4):347−53(1996))。前記研究で、平均3.9ヶ月の生存率で27%の臨床効果応答率を示した。
【0010】
2つの無作為化研究(657患者)のデーターから、局所的に進行したまたは転移NSLC患者の第一選択治療のためにゲムシタビンとシスプラチンを併用することが裏付けられている。第1の研究でゲムシタビン+シスプラチンとシスプラチン単独を比較し、第2の研究でゲムシタピン+シスプラチンとエトポシド+シスプラチンを比較した。第1の研究では全部で522被験者を対象とした(P.L.Mitchell,「クオリティオブライフ及びシスプラチン−ゲムシタビン化学療法(Quality Of Life And Cisplatin-Gemcitabine Chemotherapy)」,J.Clin.Oncol.,18(14):2791−2(2000))。ゲムシタビン(1000mg/m2)を28日周期の1日目、8日目及び15日目に投与し、シスプラチン(100mg/m2)は各周期の1日目に投与した。ゲムシタビン+シスプラチン治療群での平均生存時間及び病気進行までの平均時間はシスプラチン単独群に比して有意に長かった。客観的応答率はゲムシタビン+シスプラチン治療群で26%であり、シスプラチン単独では10%であった。第2の多施設研究では、IIIB期またはIV期のNSCLCの135患者に対して21日周期の1日目及び8日目にゲムシタビン(1250mg/m2)と1日目にシスプラチン(100mg/m2)、または21日周期の1日目、2日目及び3日目にエトポシド(100mg/m2)静注と1日目にシスプラチン(100mg/m2)を用いて治療した(F.Cardenalら,「局所的に進行したまたは転移性非小細胞肺癌の治療におけるゲムシタビン−シスプラチン対エトポシド−シスプラチンの無作為化III相研究(Randomized Phase III Study Of Gemcitabine-Cisplatin Versus Etoposide-Cisplatin In The Treatment Of Locally Advanced Or Metastatic Non-Small-Cell Lung Cancer)」,J.Clin.Oncol.,17(1):12−18(1999))。2つの治療群で生存率に有意な差はなかった。しかしながら、ゲムシタビン+シスプラチン治療群の病気進行までの平均時間及び客観的応答率がエトポシド+シスプラチン治療群に比して有意に長かった。
【0011】
1997年以来、ゲムシタビン治療に関連する急性呼吸窮迫症候群(ARDS)が数例報告されている。これらの症例は有意な罹患率及び死亡率を伴っている(Sabria−Triasら,Rev.Mal.Respir.,19:645−7(2002);Guptaら,Am.J.Clin.Oncol.,25(1):96−100(2002))。ゲムシタビンの肺毒性はゲムシタビン誘導全身性毛細管漏出症候群(SCLS)の検出に関連している。ゲムシタビン誘導SCLSは急速に進行する浮腫、体重増加、低血圧、血液濃縮及び低蛋白血症により特徴づけられ、血管内から間質スペースへの血漿の急速管外遊出を伴う突然の可逆的毛細管透過性亢進に起因する高い死亡率を有するまれな疾患である。最近の証拠で、ゲムシタビンSCLSはゲムシタビンの肺毒性に関する病原メカニズムであることが示唆されている(DePasoら,Ann.Oncol.,12(11):1651−2(2001))。更に、ゲムシタビンの効果及び安全性は用量よりもスケジュールにより強く依存していることも報告されている(Vermorkenら,Br.J.Cancer,76(11):1489−93(1997))。
【0012】
ゲムシタビンは抗癌剤として開発されたが、抗ウイルス剤としてのゲムシタビンはほとんど熱心に研究されていなかった。なぜならば、(i)ゲムシタビンを抗腫瘍剤として熟知している人々は、ゲムシタビンは非常に有毒なので、典型的には、週1回3〜4週間投与した後「休止週間」という投薬計画に従ってしか通常投与されないことを知っているから(下表1参照)、及び(ii)標準の抗ウイルス治療はヌクレオシド類縁体を無期限に、多分患者の一生の間毎日投与することからなるからである。
【0013】
【表1】
【0014】
【表2】
【0015】
表2の精査から、抗ウイルス剤治療はウイルス量の1〜2log低下を維持するために長期間にわたり毎日投与する必要があることが示される。通常、ウイルス学者は、抗ウイルス剤の治療を中止した(または、時々しか投与しなかった)場合にウイルスが消滅されていなかったならば、ウイルス量は急速にリバウンドし、持続的治療効果が得られないことを認めている。
【0016】
1986年にLilly Research LaboratoriesのDelongらは、
「2’,2’−ジフルオロ−2’−デオキシリボースを含有するヌクレオシド群を合成してその抗ウイルス活性を試験できた。前形成単層において毒性を呈することなくRNA及びDNAウイルスの両方に対して非常に高いインビトロ活性を有しているシチジンアナログが特に興味深い。この化合物は、チミジンキナーゼネガティブであり、改変DNAポリメラーゼを有するFMAU及びアシクログアノシンに対して耐性のHSV−1変異体をも阻害した。毒性は培養中の急速に増殖する細胞で観察された。前記化合物の抗ウイルス効果について各種動物モデルで試験した。化合物は動物の急性ウイルス感染においてウイルス増加を抑制したが、我々は毒性とウイルス活性を区別することはできなかった。しかしながら、我々は、マウスのフレンド白血病ウイルス感染において投薬スケジュールを変えることにより毒性と区別できる非常に高い活性を観察した。脾臓肥大及び多赤芽球症は体重が正常に増加する条件で90%抑制できた。5日毎に治療を要する投薬スケジュールが可能であった。経口及びIP経路で活性が観察された。感染のために脾臓が肥大したマウスの脾臓を治療により縮小できたことを示す研究を実施した。」
という抄録を発表した。
【0017】
強調は、追記した。米国微生物学会の年次総会の抄録(1986;Abstract No.T−56)。著者はウイルス感染(フレンド白血病ウイルス)の1症例で活性を毒性と区別できたと報告したが、これは明らかに毒性と活性を区別できないことを示す報告パターンの孤立した例外である。
【0018】
米国特許第5,015,743号明細書は、ウイルス疾患の治療のためのゲムシタビンを含めた2,2−ジフルオロ−2−デスオキシ炭水化物ヌクレオシドの属を開示している。この特許文献は、「この発明の抗ウイルス性ヌクレオシドはウイルス感染の治療のためにこの病気の治療における通常の方法で使用される」と教示している。実際、ゲムシタビンは一般的な抗ウイルス治療に従って毎日無期限に投与することはできないことは現在公知である。この特許文献はインビトロ生物学的活性の実施例を1つ含んでいるが、この“テスト1”は試験した化合物を明確に同定していない。毒性を調べるインビボデータは提示されていない。
【0019】
Hoffmann−LaRoche社が出願した国際特許出願公開第02/18404号パンフレット及び米国特許出願公開第2003/0008841号明細書は、C型肝炎を治療するための特定ヌクレオシド誘導体を開示している。ゲムシタビンはこの出願の表1、実施例243に示されている化合物243である。投薬に関して、Rocheの明細書は、
「C型肝炎ウイルス感染の治療のために必要な式Iを有する化合物の量は病気の重篤度、レシピエントの種類、性別及び体重を含めた多数の要因に依存し、最終的には担当医の裁量にまかされる。しかしながら、通常適当な有効量は0.05〜100mg/レシピエントの体重kg/日の範囲であり、好ましくは0.1〜50mg/体重kg/日の範囲、最も好ましくは0.5〜20mg/体重kg/日の範囲である。最適用量は約2〜16mg/体重kg/日である。所望用量を1日を通して適当な間隔で2回、3回、4回、5回、6回またはそれ以上に分割して投与することが好ましい。分割量は1剤形あたり例えば1〜1500mg、好ましくは5〜1000mg、最も好ましくは10〜700mgの活性成分を含む1回服用量剤形で投与され得る。」
と教示している。
【0020】
繰り返すと、ウイルス感染を治療するためにはゲムシタビンを含む上記化合物を1日数回ではないとしても、毎日用いなければならないと公衆は教示されている。毒性が公知であるので、少なくともゲムシタビンに関するこの教示は「死細胞は生存ウイルスを含まない」との古い格言の範疇に入ると見られる。しかしながら、まともな医者はウイルス感染を治療するための手段として慢性薬物中毒のために患者を殺すかまたは患者に対して重大なダメージを与えることはないであろう。従って、従来の報告に関係なく、HCVを含めたフラビウイルス感染を治療するためにゲムシタビンを使用することを誰も真剣に検討していない。
【0021】
米国特許出願公開第2002/0052317号明細書及び国際特許出願公開第02/10743号パンフレットは、インターフェロンに対する耐性を改良するためにエリスロポエチンを使用し、この療法では場合によりゲムシタビンを含むヌクレオシド類縁体の上位概念のクラスの1つを投与してもよいことを開示している。
【0022】
C型肝炎ウイルスを含むフラビウイルス科
フラビウイルスは9〜15kbのゲノムイズを有する正の一本鎖RNAウイルス群である。これらのウイルスは約45〜50nmのエンベロープウイルスである。フラビウイルス分類学の概説はウイルス分類学国際委員会から入手可能である。フラビウイルス科は3つの属からなる。
【0023】
1. フラビウイルス
この属には、デング熱ウイルス群(デング熱ウイルス、デング熱ウイルスタイプ1、デング熱ウイルスタイプ2、デング熱ウイルスタイプ3、デング熱ウイルスタイプ4)、日本脳炎ウイルス群(Alfuyウイルス、日本脳炎ウイルス、Kookaburraウイルス、Koutangoウイルス、クンジンウイルス、マレー渓谷脳炎ウイルス、セントルイス脳炎ウイルス、ストラットフォードウイルス、Usutuウイルス、西ナイルウイルス)、モドックウイルス群、リオブラボウイルス群(アポイウイルス、リオブラボウイルス、サボヤウイルス)、ヌタヤ(Ntaya)ウイルス群、マダニ伝搬脳炎ウイルス群(マダニ伝搬脳炎ウイルス)、Tyuleniyウイルス群、ウガンダSウイルス群及び黄熱ウイルス群が含まれる。これらの主要群の他に、分類されていない幾つかの追加フラビウイルスが存在する。
【0024】
2. ペスチウイルス
この属には、ウシウイルス性下痢症ウイルス−2(BVDV−2)、(BVDVを含めた)ペスチウイルスタイプ1、(豚コレラウイルスを含めた)ペスチウイルスタイプ2及び(ボーダー病ウイルスを含めた)ペスチウイルス3が含まれる。
【0025】
3. 肝炎ウイルス
この属には1つの種のみ、すなわち多くのクレード、タイプ及びサブタイプから構成されるC型肝炎ウイルス(HCV)のみが含まれる。
【0026】
HCVは1989年まで同定されておらず、それまで非A非B肝炎と称されていた。HCVはB型肝炎と合わせて世界中の肝疾患のすべての症例の75%を占めている(B.Helblingら,「未処置の慢性C型肝炎におけるインターフェロン及びアマンタジン:プラセボをコントロールとする二重盲検無作為化治験(Interferon And Amantadine In Naive Chronic Hepatitis C: A Doubleblind, Randomized, Placebo-Controlled Trial)」,Hepatology,35(2):447−54(2002))。HCV感染に関連する肝不全は米国での肝移植の主な原因である。HCV感染は通常初期段階では軽度であるので、慢性段階になるまで診断されにくい。従って、HCVはしばしば「無症候流行病」と呼ばれている。HCVは感染から肝疾患の発症までの典型型サイクルには約20年を要し、よって増えつつある感染者に対するこの病気の真の影響は多年にわたり現れない。HCVは感染者の血液と接触することにより広がる。HCV感染のリスク因子が高い人には、非合法注射薬の使用者、1992年以前に輸血または固形臓器移植を受けた人、1987年以前に凝血の問題のために血液製剤を投与された人、長期間腎臓透析を受けている患者、肝疾患の証拠を呈している人(例えば、ALTレベルが持続的に異常な人)が含まれる。
【0027】
米国では約400万人がHCVに感染しており、世界中では2億人以上が感染していると推定される(S.E.Hewitt,「C型肝炎ウイルス(HCV)感染及びHCV関連疾患の予防及びコントロールのための推奨(Recommendations for Prevention and Control of Hepatitis C Virus (HCV) infection and HCV-related Disease)」,疾病管理及び予防センター(1998))。1980年代では毎年平均230,000人が新たに感染している。1989年以降、新たに感染した患者の数は1986年までの>80%減少し、36,000人になった。これらの患者の多くは慢性的に感染しており、症状が出ないままであるのでこの感染に気づいていない場合がある。よって、HCV関連肝疾患は今世紀に直面している最大の公衆衛生脅威の1つであり得る。
【0028】
慢性肝疾患は米国の成人の死亡の主因の10位であり、毎年25,000人が死亡している。集団研究で、慢性肝疾患の40%がHCVに関連していると推定される。多くのHCV感染者は30〜49歳であるので、HCV関連の慢性肝疾患による死亡者の数は次の10〜20年に実質的に増加する恐れがある。HCV関連合併症の現在の医療費が>6億ドル/年と推定されているので、これは軽微なことでない。
【0029】
HCVはRNAウイルスであり、これは頻繁に変異することを意味する(www.epidemic.org/index2.html,「C型肝炎に関する事実(The Facts about Hepatitis C)」,1998,ダートマス大学)。一旦感染すると、HCVは宿主の体内でそれ自体の異なった遺伝的変異を生ずる。変異体はしばしばその前駆体とは異なり、免疫系は変異体を認識できない。よって、たとえ免疫系が1つの変異に対して成功しても、変異株はすぐに支配的になり、優勢株となる。このことが、HCV感染者の>80%が慢性肝疾患に進行する理由となる。HCVは6つの主要遺伝子型及び50以上のサブタイプを有する。米国では、HCV感染患者の中で約70%が遺伝子型1を有し、15%が遺伝子型2を有し、10%が遺伝子型3を有する(J.G.McHutchisonら,「慢性C型肝炎の初期治療としてのインターフェロン Alfa−2b単独とリバビリンとの併用(Interferon Alfa-2b Alone Or In Combination With Ribavirin As Initial Treatment For Chronic Hepatitis C)」,Hepatitis Interventional Therapy Group,N.Engl.J.Med.,339(21):1485−92(1998))。肝硬変への進行のリスクを有する慢性HCV感染患者に対して抗ウイルス治療が推奨されている(S.K.Herrine,「慢性C型肝炎ウイルス感染患者に対するアプローチ(Approach To The Patient With Chronic Hepatitis C Virus Infection)」,Ann.Intern.Med.,136(10):747−57(2002))。上記した患者には、ALTレベルが持続的に高く、HCV RNAが検出可能であり、肝生検で門脈または架橋状線維症を示したかまたは少なくとも軽度の炎症または壊死を示している抗−HCV抗体陽性患者が含まれる。
【0030】
HCVに対する治療は急速に変化しており、インターフェロン、リバビリンやヌクレオシド類縁体との併用治療が慢性HCV感染を有する未処置患者を治療するために米国で認可されている(S.E.Hewitt,「C型肝炎ウイルス(HCV)感染及びHCV関連疾患の予防及びコントロールのための推奨(Recommentations for Prevention and Control of Hepatitis C Virus (HCV) infection and HCV-related Disease)」,疾病管理及び予防センター(1998))。持続的応答率はインターフェロン単独で治療した患者では15〜25%でしかなかったのに対して、リバビリン+インターフェロンで治療した患者では40〜50%に達した。しかしながら、米国で最も一般的なHCV遺伝子型である遺伝子型1を有する患者での併用治療の成功率は低く、これらの患者での持続的応答率は<30%にすぎない。更に、治療に伴う副作用によりしばしば治療の用量を低下させたり、治療期間を中断させている。インターフェロン+リバビリン治療に伴ってしばしば生ずる副作用にはインフルエンザ様症候群、過敏症、うつ病、貧血、骨髄抑制及び腎不全が含まれる。リバビリンは妊娠の可能性ある女性では催奇形性であり、禁忌である。
【0031】
慢性HCV感染による公衆衛生の脅威及び現在の治療の限界のために、HCV感染を治療するための革新的治療アプローチが要望されている。
【発明の開示】
【0032】
従って、本発明の目的はフラビウイルス、特にHCV感染を治療するための新規組成物及び方法を提供することである。
【0033】
驚くべきことに、最少用量のゲムシタビン(または、本明細書に記載されているその塩、プロドラッグもしくは誘導体)によりヒト患者におけるC型肝炎のウイルス量を数日間未満、実際ある場合には1〜2日間以下で最高2logまで、またはそれ以上低下させることができることが知見された。こうしたウイルス量の急速で大きな低下は、約14日以上毎日持続的に治療した後に1〜2log安定的にしか低下できない従来の抗ウイルス治療経験と対照的である。ヒトにおけるゲムシタビン、またはその塩もしくはプロドラッグの予期しない強くユニークな抗−HCV活性により抗ウイルス薬物投与モデルの基本的変更の基礎が与えられ、前記治療のためにゲムシタビンを保存的にかつ適切に使用することが初めてできた。
【0034】
従って、第1実施態様において、本発明は、ゲムシタビン(または、本明細書に記載されているその塩、プロドラッグもしくは誘導体)を約50〜約1300mg/m2/日の範囲用量で1、2又は3日間投与した後治療を休止することを含むフラビウイルス感染、特にC型肝炎ウイルス感染を治療するための方法及び組成物を提供する。場合により、ウイルス量を経時的にモニターしてウイルスリバウンドを調べる。有意なウイルス量が再び観察されない限り治療を再開せず、観察されたときに1、2又は3日間の治療を繰り返す。この治療は患者の健康をモニターし、維持するために無期限に継続することができる。
【0035】
治療され得るフラビウイルス科ウイルスには、肝炎ウイルス属(HCV)、ペスチウイルス属(BVDV、CSFV、BDV)及びフラビウイルス属(デング熱ウイルス、西ナイルウイルスを含めた日本脳炎ウイルス群、及び黄熱ウイルス)のすべてのメンバーが含まれる。
【0036】
別の実施態様では、より重篤なフラビウイルス科感染に対してゲムシタビン(または、本明細書に記載されているその塩、プロドラッグもしくは誘導体)を約50〜約1300mg/m2/日の範囲用量で1日間〜7日間(例えば、1、2、3、4、5、6または7日間)投与した後治療を休止する。場合により、ウイルス量を経時的にモニターし、休止後のウイルスリバウンドをモニターする。有意なウイルス量が再び観察されない限り治療を再開せず、観察されたときに1〜7日間(例えば、独立して、1、2、3、4、5、6または7日間)、より好ましくは1、2又は3日間の治療は繰り返す。この治療は患者の健康をモニターし、維持するために無期限に継続することができる。
【0037】
本発明はゲムシタビン、その塩もしくはプロドラッグを用いて抗ウイルス治療を抗腫瘍投薬スケジュールを用いて実施し得ることを初めて開示している。特定実施態様では、フラビウイルス感染を治療するためにゲムシタビンに対する認可されたいずれもの抗腫瘍投薬スケジュールを使用することができる。
【0038】
幾つかの非限定的例示実施態様では、ゲムシタビンの1日用量は100〜1500mg/日、または200〜1000mg/日、特に300〜800mg/日の範囲であり得る。
【0039】
1つの例示実施態様では、1日目に患者に静注投与し、薬物投与から数時間、多分最高12時間病院に留まるよう指示する。患者を安全性及び耐性についてモニターし、HCV−RNAを調べるために投与前、投与から6時間目及び12時間目に採血する。2日目には安全性評価及びウイルス量測定のために患者に再び来院させる。場合により治療を2、3、4、5、6及び7日目にも継続する。その後、治療を休止し、患者には安全性及びウイルス量試験を追跡するために定期的に来院するように指示する。
【0040】
ゲムシタビンは消化管で急速にそのウラシル誘導体に変換されることが公知であるので、ゲムシタビンを静注の形態で投与することが好ましい。ゲムシタビンを経口投与することが好ましいならば、前記化合物をプロドラッグであって、活性に悪影響を与えることなくシトシニルアミン基が急速脱アミノ化されることを保護するプロドラッグの形態で投与することが好ましい。シトシン塩基のインビボでの半減期を延長させるための非限定的方法として、前記化合物をN−アシル化、N−アルキル化またはN−アリール化形態で投与することが挙げられる。
【0041】
プロドラッグには、本明細書に記載のヌクレオシドのエステル及びアミドを形成するためのヒドロキシまたはアミノ官能基上のアミノ酸誘導体も含まれる。例えば、アシクロビルそのものに比較して改良された水溶性を有するアシクロビルのアミノ酸エステル、特にグリシンエステル及びアラニンエステルを開示している欧州特許出願公開第99493号明細書、及びアシクロビルのアラニンエステル及びグリシンエステルに比較して経口投与後高いバイオアベイラビリティを示すα−炭素原子に隣接した側鎖分枝鎖を特徴とするアシクロビルのバリンエステルを開示している米国特許第4,957,924号明細書(Beauchamp)を参照されたい。前記アミノ酸エステルの製造方法は米国特許第4,957,924号明細書(Beauchamp)に記載されている。バリンそのものの使用に代えて、アミノ酸の機能的均等物(例えば、酸クロリドのような酸ハライドまたは酸無水物)を使用してもよい。その場合、望ましくない副作用を避けるために、アミノ保護誘導体を使用することが有利であり得る。
【0042】
本発明の1例として、HCVに感染した多病巣性HCC、肝硬変及び虚血性肝炎を呈している男性患者にゲムシタビンHCl 1200mgを1000分間かけてオキサリプラチンと一緒に投与した。耐性は許容でき、よって翌日患者には再びゲムシタビン約700mgを投与した。第2回投与前のベースラインのウイルス量は6.49logコピー/mlであった。心臓に問題が生じたので、約700mgを投与後ゲムシタビンの2回目の注入を停止した。心臓の問題は明らかにゲムシタビン治療とは関係なかった。第2回投与から8時間後のHCV RNA測定値は4.04logコピー/mlであった。8時間で約2.5logの低下を示した。
【0043】
別の実施態様では、ゲムシタビン、またはその塩、プロドラッグもしくは誘導体を1つ以上の他の抗−フラビウイルス活性剤と一緒にまたは交互に本明細書に記載の投薬計画に従って投与する。以下に詳記する他の活性剤はこの投薬計画と組み合わせてその効果を最大限とするように投与される。
【発明を実施するための最良の形態】
【0044】
驚くべきことに、最少量のゲムシタビンが数日間未満で、実際にはある場合には1〜2日間以内でヒト患者においてC型肝炎のウイルス量を最高2logまで、またはそれ以上低下させることができることが知見された。このようにウイルス量が急速に大きく低下することは、1〜2logの低下が約14日間以上毎日継続治療した後に安定的にしか達成されない従来の抗ウイルス経験と対照的である。ヒトにおけるゲムシタビンの予期せぬ強くユニークな抗−HCV活性により、抗ウイルス薬の投薬モデルの基本的変更の基礎が与えられ、初めて前記治療のために前記薬物が保存的にかつ適切に使用できた。
【0045】
従って、第1態様で、本発明は、ゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグを約50〜約1300mg/m2/日の範囲の用量で1、2又は3日間投与した後治療を休止することを含むフラビウイルス感染、特にC型肝炎ウイルス感染の治療のための方法及び組成物を提供する。場合により、ウイルス量を経時的にモニターしてウイルスリバウンドを調べてもよい。有意なウイルス量が再び観察されない限り治療を再開せず、観察されたときには1、2又は3日間の治療を繰り返す。この治療は患者の健康をモニターし、維持するために無期限に継続され得る。
【0046】
治療され得るフラビウイルスには、肝炎ウイルス属(HCV)、ペスチウイルス属(BVDV、CSFV、BDV)及びフラビウイルス属(デング熱ウイルス、西ナイルウイルスを含めた日本脳炎ウイルス群及び黄熱ウイルス)のすべてのメンバーが含まれ得る。
【0047】
別の実施態様において、より重篤なフラビウイルス感染の場合、ゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグを約50〜約1300mg/m2/日の範囲の用量で1日〜7日間投与した後治療を休止する。場合によりウイルス量を経時的にモニターしてウイルスのリバウンドを調べてもよい。有意なウイルス量が再び観察されない限り治療を再開せず、観察されたときには1〜7日間、より好ましくは1、2又は3日間の治療を繰り返す。この治療は患者の健康をモニターし、維持するために無期限に継続され得る。
【0048】
I.本発明の化合物
本発明の特定実施態様において、式:
【0049】
【化5】
を有するβ−D−ヌクレオシド、そのβ−L−エナンチオマー、その医薬的に許容され得る塩もしくはプロドラッグがフラビウイルス感染、特にHCV感染を治療または予防するために提供される。好ましい実施態様において、化合物はゲムシタビン、またはその医薬的に許容され得る塩、エステルもしくはプロドラッグである。前記化合物は、例えばその活性、バイオアベイラビリティ、安定性を修飾するため、またはヌクレオシドの諸特性を変化させるためにN4及び/または3’位及び/または5’位でアルキル化、アシル化または他の方法で誘導体化されていてもよい。こうすると、非静脈処方物をより安定性とし得る。1つの実施態様において、化合物はN4及び/または3’位及び/または5’位をバリンのようなアミノ酸でアシル化されている。
【0050】
本発明の包括的な側面において、活性化合物は一般式(I)を有するβ−Dもしくはβ−L−ヌクレオシド、またはその医薬的に許容され得る塩もしくはプロドラッグ(本明細書中、ゲムシタビン誘導体と称する)である。
【0051】
【化6】
【0052】
上記式中、
RはH、ハロゲン(F、Cl、Br、I)、OH、OR’、SH、SR’、NH2、NHR’、NR’2、C1−6低級アルキル、ハロゲン化(F、Cl、Br、I)C1−6低級アルキル(例えば、CF3及びCH2CH2F)、C2−6低級アルケニル(例えば、CH=CH2)、ハロゲン化(F、Cl、Br、I)C2−6低級アルケニル(例えば、CH=CHCl、CH=CHBr及びCH=CHI)、C2−6低級アルキニル(例えば、C≡CH)、ハロゲン化(F、Cl、Br、I)C2−6低級アルキニル、C1−6低級アルコキシ(例えば、CH2OH及びCH2CH2OH)、CO2H、CO2R’、CONH2、CONHR’、CONR’2、CH=CHCO2H、CH=CHCO2R’であり;
XはH、ハロゲン、OH、OR’、OCH3、SH、SR’、SCH3、NH2、NHR’、NR’2またはCH3であり;
各R’は独立して水素、C1−6低級アルキルまたはC1−6低級シクロアルキルであり;
ZはO、SまたはCH2であり;
R4はH、モノホスフェート、ジホスフェート、トリホスフェート、安定化ホスフェートプロドラッグ、アシル、アルキル、スルホネートエステル、脂質、リン脂質、アミノ酸、炭水化物、ペプチド、コレステロール、またはインビボ投与したときにR4がHまたはホスフェートである化合物を生じ得る他の医薬的に許容され得る離脱基であり、;
R3はFまたはOHである。
【0053】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはハロゲンである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0054】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはアルキルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0055】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはハロゲン化アルキルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0056】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはCH3であり、RはNH2である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0057】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはOR’であり、Rはハロゲンである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0058】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはハロゲンであり、R4は水素である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0059】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0060】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはアルケニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0061】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはアルキニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0062】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはハロゲン化アルケニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0063】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはハロゲン化アルキニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0064】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはアルコキシである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0065】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、RはCO2Hである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0066】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、RはCO2R’である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0067】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、RはCONH2である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0068】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、RはCONHR’である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0069】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはハロゲンである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0070】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、Rはハロゲンである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0071】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、Rはアルキルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0072】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、Rはハロゲン化アルキルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0073】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはCH3であり、R3はOHであり、RはNH2である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0074】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはOR’であり、R3はOHであり、Rはハロゲンである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0075】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、Rはハロゲンであり、R4は水素である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0076】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0077】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはCH3であり、R3はFであり、Rはアルケニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0078】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはCH3であり、R3はOHであり、Rはアルキニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0079】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、Rはハロゲン化アルケニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0080】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、Rはハロゲン化アルキニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0081】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、Rはアルコキシである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0082】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、RはCO2Hである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0083】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、RはCO2R’である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0084】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、RはCONH2である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0085】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、RはCONHR’である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0086】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、Rはハロゲンである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0087】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、Rはハロゲンであり、R4はHである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0088】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはSH2であり、R3はOHであり、Rはハロゲンである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0089】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNHR’であり、R3はOHであり、Rはハロゲンである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0090】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、Rはハロゲンである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0091】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、Rはアルキルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0092】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、Rはアルケニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0093】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、Rはアルキニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0094】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、Rはハロゲン化アルケニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0095】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、Rはハロゲン化アルキニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0096】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、Rはアルコキシである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0097】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、RはCO2Hである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0098】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはOR’である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0099】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNHR’である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0100】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはCONH2である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0101】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R3はFである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0102】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R3はOHである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0103】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R4はHである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0104】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R4はモノホスフェートである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0105】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R4はジホスフェートである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0106】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R4はトリホスフェートである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0107】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R4はアシルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0108】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R4はHであり、ZはOである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0109】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R4はHであり、ZはCH2である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0110】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、RはHであり、R3はFであり、R4はアシルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0111】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R4はHであり、RはOR’である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0112】
定義
本明細書中、用語「アルキル」は、別段のことわりがない限り飽和の直鎖、分枝鎖または環状の1級、2級または3級炭化水素を指し、この中には非限定的にC1−16アルキルが含まれる。具体的には、メチル、エチル、プロピル、イソプロピル、シクロプロピル、ブチル、イソブチル、t−ブチル、ペンチル、シクロペンチル、イソペンチル、ネオペンチル、ヘキシル、イソヘキシル、シクロヘキシル、シクロヘキシルメチル、3−メチルペンチル、2,2−ジメチルブチル及び2,3−ジメチルブチルが含まれる。アルキル基は場合により、保護されていなくても所要により保護されていてもよい、アルキル、ハロ、ハロアルキル、ヒドロキシ、カルボキシル、アシル、アシルオキシ、アミノ、アミド(amido)、カルボキシル誘導体、アルキルアミノ、ジアルキルアミノ、アリールアミノ、アルコキシ、アリールオキシ、ニトロ、シアノ、アジド、チオール、イミン、スルホン酸、スルフェート、スルホニル、スルファニル、スルフィニル、スルファモイル、エステル、カルボン酸、アミド(amide)、ホスホニル、ホスフィニル、ホスホリル、ホスフィン、チオエステル、チオエーテル、酸ハライド、無水物、オキシム、ヒドロジン、カルバメート、ホスホン酸、ホスフェート、ホスホネート、及び本化合物の医薬活性を阻害しない任意の実行可能な官能基からなる群から選択される1つ以上の部分で置換されていてもよい。なお、保護基は援用により本明細書に含まれるとするGreeneら,「有機合成における保護基(Protective Groups in Organic Synthesis)」,第2版,John Wiley and Sons(1991年)発行に教示されているように当業者に公知である。
【0113】
本明細書中、用語「低級アルキル」は、別段のことわりがない限りC1−4飽和直鎖、分枝鎖、または適当ならば環状(例えば、シクロプロピル)アルキル基を指し、この基は未置換でも置換されていてもよい。
【0114】
本明細書中、用語「アルキレン」または「アルケニル」は、直鎖または分枝鎖構造を有する飽和ヒドロカルビルジイル基を指し、この中には非限定的に1〜10炭素原子を有するものが含まれる。この用語の範囲には、メチレン、1,2−エタン−ジイル、1,1−エタン−ジイル、1,3−プロパン−ジイル、1,2−プロパン−ジイル、1,3−ブタン−ジイル、1,4−ブタン−ジイル等が含まれる。アルキレン基または本明細書に記載されている他の2価部分は場合により、保護されていなくても所要により保護されていてもよい、アルキル、ハロ、ハロアルキル、ヒドロキシ、カルボキシル、アシル、アシルオキシ、アミノ、アミド、カルボキシル誘導体、アルキルアミノ、アジド、ジアルキルアミノ、アリールアミノ、アルコキシ、アリールオキシ、ニトロ、シアノ、スルホン酸、チオール、イミン、スルホニル、スルファニル、スルフィニル、スルファモイル、エステル、カルボン酸、アミド、ホスホニル、ホスフィニル、ホスホリル、ホスフィン、チオエステル、チオエーテル、酸ハライド、無水物、オキシム、ヒドロジン、カルバメート、ホスホン酸、ホスホネート、及び本化合物の医薬活性を阻害しない任意の実行可能な官能基からなる群から選択される1つ以上の部分で置換されていてもよい。なお、保護基は援用により本明細書に含まれるとするGreeneら,「有機合成における保護基(Protective Groups in Organic Synthesis)」,第2版,John Wiley and Sons(1991年)発行に教示されているように当業者に公知である。
【0115】
本明細書中、用語「アリール」は、別段のことわりがない限りフェニル、ビフェニルまたはナフチルを指し、好ましくはフェニルである。この用語は置換部分及び未置換部分を含む。アリール基は、保護されていなくても所要により保護されていてもよい、ブロモ、クロロ、フルオロ、ヨード、ヒドロキシ、アジド、アミノ、アルキルアミノ、アリールアミノ、アルコキシ、アリールオキシ、ニトロ、シアノ、スルホン酸、スルフェート、ホスホン酸、ホスフェート及びホスホネートからなる群から選択される1つ以上の部分で置換されていてもよい。なお、保護基は援用により本明細書に含まれるとするGreeneら,「有機合成における保護基(Protective Groups in Organic Synthesis)」,第2版,John Wiley and Sons(1991年)発行に教示されているように当業者に公知である。
【0116】
本明細書中、用語「アミノ酸」は、天然及び合成α、β、γまたはδ−アミノ酸を含み、その中には非限定的にアラニル、バリニル、ロイシニル、イソロイシニル、プロリニル、フェニルアラニニル、トリプトファニル、メチオニニル、グリシニル、セリニル、スレオニニル、システイニル、チロシニル、アスパラギニル、グルタミニル、アスパルトイル、グルタロイル、リシニル、アルギニニル、ヒスチジニル、β−アラニル、β−バリニル、β−ロイシニル、β−イソロイシニル、β−プロリニル、β−フェニルアラニニル、β−トリプトファニル、β−メチオニニル、β−グリシニル、β−セリニル、β−スレオニニル、β−システイニル、β−チロシニル、β−アスパラギニル、β−グルタミニル、β−アスパルトイル、β−グルタロイル、β−リシニル、β−アラギニニル及びβ−ヒスチジニルが含まれる。
【0117】
本明細書中、用語「アラルキル」は、別段のことわりがない限り上記アルキル基を介して分子に結合した上記アリール基を指す。本明細書中、用語「アルカリール」または「アルキルアリール」は、別段のことわりがない限り上記アリール基を介して分子に結合した上記アルキル基を指す。これらの各基においてアルキル基は場合により上記したように置換されていてもよく、アリール基は場合により、保護されていなくても所要により保護されていてもよい、アルキル、ハロ、ハロアルキル、ヒドロキシ、カルボキシル、アシル、アシルオキシ、アミノ、アミド、アジド、カルボキシル誘導体、アルキルアミノ、ジアルキルアミノ、アリールアミノ、アルコキシ、アリールオキシ、ニトロ、シアノ、スルホン酸、チオール、イミン、スルホニル、スルファニル、スルフィニル、スルファモイル、エステル、カルボン酸、アミド、ホスホニル、ホスフィニル、ホスホリル、ホスフィン、チオエステル、チオエーテル、酸ハライド、無水物、オキシム、ヒドロジン、カルバメート、ホスホン酸、ホスホネート、及び本化合物の医薬活性を阻害しない任意の実行可能な官能基からなる群から選択される1つ以上の部分で置換されていてもよい。なお、保護基は援用により本明細書に含まれるとするGreeneら,「有機合成における保護基(Protective Groups in Organic Synthesis)」,第2版,John Wiley and Sons(1991年)発行に教示されているように当業者に公知である。用語「アリール」の範囲内には、具体的にフェニル、ナフチル、フェニルメチル、フェニルエチル、3,4,5−トリヒドロキシフェニル、3,4,5−トリメトキシフェニル、3,4,5−トリエトキシフェニル、4−クロロフェニル、4−メチルフェニル、3,5−ジ−t−ブチル−4−ヒドロキシフェニル、4−フルオロフェニル、4−クロロ−1−ナフチル、2−メチル−1−ナフチルメチル、2−ナフチルメチル、4−クロロフェニルメチル、4−t−ブチルフェニル、4−t−ブチルフェニルメチル等が含まれる。
【0118】
用語「アルキルアミノ」または「アリールアミノ」は、それぞれ1または2個のアルキル置換基またはアリール置換基を有するアミノ基を指す。
【0119】
本明細書中、用語「ハロゲン」は、フッ素、塩素、臭素及びヨウ素を含む。
【0120】
用語「エニンチオマー的に富化された」は、本明細書中、ヌクレオシドであって、当該ヌクレオシドの単一エナンチオマーを少なくとも約95%、好ましくは少なくとも96%、より好ましくは少なくとも97%、更に好ましくは少なくとも98%、特に好ましくは少なくとも約99%以上含むものを指すべく使用される。好ましい実施態様において、ヌクレオシド類縁体はエナンチオマー的に富化された形態で提供される。
【0121】
本明細書中、用語「宿主」は、その中でウイルスが複製し得る単細胞または多細胞生物を指し、この中には細胞株及び動物が含まれ、好ましくはヒトである。或いは、宿主は複製または機能が本発明の化合物により変化を受け得るウイルスゲノムの一部を有していてもよい。用語「宿主」は具体的には、感染細胞、ウイルスゲノムの全部または一部をトランスフェクトした細胞及び動物、特に(チンパンジーを含めた)霊長類及びヒトを指す。異常細胞増殖に関して、用語「宿主」は異常細胞増殖を模倣し得る単細胞または多細胞生物を指す。用語「宿主」は具体的には、自然または不自然な原因により(例えば、それぞれ遺伝的変異または遺伝子工学操作により)異常に増殖する細胞及び動物、特に(チンパンジーを含めた)霊長類及びヒトを指す。本発明の多くの動物の用途において、宿主はヒト患者である。しかしながら、獣医学的用途(例えば、ウシにおけるウシウイルス性下痢症ウイルス、豚の豚コレラウイルス及びヒツジのボーダー病ウイルス)は本発明により明らかに予測される。
【0122】
用語「医薬的に許容され得る塩もしくはプロドラッグ」は、本明細書中、患者に投与したときに活性化合物を与える化合物の医薬的に許容され得るいずれもの形態(例えば、エステル、ホスフェートエステル、エステルまたは関連基の塩)を指すべく使用される。医薬的に許容され得る塩には、医薬的に許容され得る無機もしくは有機塩基または無機もしくは有機酸から誘導されるものが含まれる。適当な塩には、アルカリ金属(例えば、カリウム及びナトリウム)及びアルカリ土類金属(例えば、カルシウム及びマグネシウム)の他、製薬業界で公知の多数の他の酸から誘導されるものが含まれる。医薬的に許容され得るプロドラッグは宿主において代謝(例えば、加水分解または酸化)されて本発明の化合物を形成する化合物を指す。プロドラッグの典型例には、活性化合物の官能基部分上に生物学的に安定な保護基を有する化合物が含まれる。プロドラッグには、酸化、還元、アミノ化、脱アミノ化、ヒドロキシル化、脱ヒドロキシル化、加水分解、脱加水分解、アルキル化、脱アルキル化、アシル化、脱アシル化、リン酸化、脱リン酸化されると活性化合物を生じさせ得る化合物が含まれる。
【0123】
本発明の化合物はフラビウイルス科ウイルスに対して抗ウイルス活性を有するかもしくは異常細胞増殖に対して抗増殖活性を有し、または前記活性を呈する化合物に代謝される。
【0124】
立体異性及び多形
本発明の化合物は少なくとも2つのキラル中心を有し、光学的に活性な形態及びラセミ体形態で存在し得、単離され得る。幾つかの化合物は多型を示し得る。本発明は、本明細書に記載されている有用な特性を有する本発明の化合物のラセミ、光学活性、多型または立体異性体形態、またはそれらの混合物を包含する。光学活性形態は、例えばラセミ体形態を再結晶化技術、光学活性な出発物質からの合成、キラル合成、またはキラル固定相を用いるクロマトグラフィー分離、または酵素分割により製造され得る。
【0125】
化合物の光学的に活性な形態は、再結晶技術によるラセミ形態の分割、光学活性出発物質からの合成、キラル合成、またはキラル固定相を用いるクロマトグラフィー分離を含めた当業界で公知の方法を用いて製造され得る。
【0126】
光学的に活性な物質を得るための方法の例は以下の手順を少なくとも1つ含む。
i) 結晶の物理的分離:
各エナンチオマーの巨視的結晶を手動で分離する技術。この技術は、別のエナンチオマーの結晶が存在する場合、すなわち物質が集塊で、結晶が視覚的に認識できる場合に使用され得る。
ii) 同時結晶化:
個々のエナンチオマーをラセミ体溶液から別々に結晶化させる技術。ラセミ体が固体の集塊のときにのみ使用され得る。
iii) 酵素分割:
エナンチオマーに対する酵素との反応速度の違いを利用してラセミ体を部分的または完全に分離させる技術。
iv) 酵素的非対称合成:
所望エナンチオマーのエナンチオマー的に純粋なまたはエナンチオマー的に富化された合成前駆体を得るために合成の少なくとも1工程が酵素反応を利用している合成技術。
v) 化学非対称合成:
所望エナンチオマーを生成物中に非対称(すなわち、キラル性)を生ずる条件下でアキラル前駆体から合成する合成技術。非対称はキラル触媒またはキラル助剤を用いて達成され得る。
vi) ジアステレオマー分離:
ラセミ化合物を各エナンチオマーをジアステレオマーに変換するエナンチオマー的に純粋な試薬(キラル助剤)と反応させる技術。その後、生じたジアステレオマーをより区別し得る構造の違いを利用してクロマトグラフィーまたは結晶化により分離し、その後キラル助剤を除去して、所望エナンチオマーを得る。
vii) 一次及び二次非対称変換:
所望エナンチオマーからジアステレオマーが優先的に溶解するようにラセミ体からのジアステレオマーが平衡化するかまたは所望エナンチオマーからのジアステレオマーの優先的結晶化が最終的に原則としてすべての物質が所望エナンチオマーから結晶性ジアステレオマーに変換されるように平衡を混乱させる技術。その後、所望エナンチオマーをジアステレオマーから遊離させる。
viii) カイネティック分解:
この技術により、エナンチオマーのカイネティック条件下でのキラルな非ラセミ試薬または触媒との反応速度の違いを利用してラセミ体を部分的または完全に分割(または、部分的に分割した化合物の更なる分割)がなされる。
ix) 非ラセミ前駆体からのエナンチオ特異的合成:
所望エナンチオマーを非キラル出発物質から得、立体化学一体性が合成中全くまたは殆ど損なわれない技術。
x) キラル液体クロマトグラフィー:
ラセミ体のエナンチオマーを固定相との相互作用の違いを利用して(キラルHPLCを介するものを含む)液体移動相で分離する技術。固定相はキラル物質から構成され得、移動相は相互作用の違いを生じさせるために追加のキラル物質を含み得る。
xi) キラルガスクロマトグラフィー:
ラセミ体を気化させ、エナンチオマーを気体移動相での固定非ラセミキラル吸着剤相を含むカラムとの相互作用の違いを利用して分離する技術。
xii) キラル溶媒を用いる抽出:
1つのエナンチオマーを特定のキラル溶媒に優先的に溶解させることによりエナンチオマーを分離する技術。
xiii) キラル膜を介する移動:
ラセミ体を薄膜バリアと接触させる技術。前記バリアにより通常2つの混和性流体(一方はラセミ体を含む)が分離され、濃度または圧力差のような駆動力により膜バリアを優先的に移動する。分離は、ラセミ体の1つのエナンチオマーのみが通過し得る膜の非ラセミ体キラル性の結果起こる。
【0127】
模擬移動床クロマトグラフィーを含めたキラルクロマトグラフィーが1実施態様で使用される。各種のキラル固定相が市販されている。
【0128】
医薬組成物
式(I)を有する化合物、またはその医薬的に許容され得る塩もしくはプロドラッグに基づく医薬組成物は、フラビウイルス科ウイルスを処理するために治療有効量を場合により医薬的に許容され得る添加剤、担体または賦形剤と組み合わせて製造され得る。治療有効量は治療しようとする感染または状態、その重篤度、使用する治療方式、使用する化合物の薬物動態及び治療する患者に応じて異なり得る。
【0129】
本発明の1つの側面で、本発明の化合物を医薬的に許容され得る担体と混合して処方することが好ましい。通常、医薬組成物を静脈内形態で投与することが好ましいが、経口、非経口、筋肉内、経皮、頬腔、皮下、坐薬または他のルートを介して投与するようにも処方され得る。静脈内及び筋肉内処方物は好ましくは滅菌食塩液の形態で投与される。特定ルートに対する各種処方物を得るために当業者は本明細書の教示の範囲内で処方物を修飾することができる。特に、所望化合物を水または他のベヒクル中により可溶性とするための修飾は例えばルーチンの修飾(塩形成、エステル化等)により容易に実施し得る。
【0130】
経口処方物のような特定の剤形では、本発明の化合物のプロドラッグ形態、特にアシル化(アセチル化等)及びエーテル誘導体、ホスフェートエステルまたは塩形態を含めたプロドラッグ形態が好ましい。当業者は活性化合物の宿主生物または患者内の標的部位へのデリバリーを促進するために本発明の化合物をプロドラッグ形態に容易に修飾する方法を認識している。当業者は、(HCVを含めた)フラビウイルス科ウイルス感染の治療において化合物の所期効果を最大限とすべく所望化合物を宿主生物または患者の標的部位にデリバリーする際に適用可能ならばプロドラッグの有利な薬物動態パラメータも利用する。
【0131】
本発明の治療上活性な処方物中に含まれる化合物の量は(HCVを含めた)フラビウイルス科ウイルス感染を治療するために有効な量である。
【0132】
活性化合物は連続(静脈内滴注)投与しても数回(例えば、1日4回、1日2回等)経口投与してもよく、投与ルートの中には、他のルートの中で経口、局所、非経口、筋肉内、静脈内、皮下、経皮(浸透強化剤を含み得る)、頬腔及び坐薬投与が含まれ得る。経口投与ルートからの化合物のバイオアベイラビリティ及び安定性を高めるために腸溶性経口錠剤も使用し得る。最も有効な剤形は選択した特定化合物の薬物動態及び患者の病気の重篤度に依存する。
【0133】
第1実施態様において、本発明はフラビウイルス科ウイルス感染、特にC型肝炎ウイルス感染を治療するための方法及び組成物を提供し、その方法はゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグもしくは誘導体を約50〜約1300mg/m2/日の範囲の用量で1、2又は3日間投与した後治療を休止することを含む。別の実施態様では、より重篤なフラビウイルス科ウイルス感染に対してゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグもしくは誘導体を約50〜約1300mg/m2/日の用量で1日〜7日間(例えば、1、2、3、4、5、6または7日間)投与した後治療を休止する。
【0134】
本発明のゲムシタビンまたは別の活性化合物の1日用量は治療効果を最大限とするように選択され得る。非限定的用量の例は100〜1500mg/日、または200〜1000mg/日、特に300〜800mg/日である。
【0135】
化合物が安定な非毒性の酸性または塩基性塩を形成するのに十分な塩基性または酸性である場合には、前記化合物を医薬的に許容され得る塩として投与することも適当であり得る。医薬的に許容され得る塩には、医薬的に許容され得る無機もしくは有機塩基及び医薬的に許容され得る無機もしくは有機酸から誘導されるものが含まれる。適当な塩には、アルカリ金属(例えば、カリウム及びナトリウム)、アルカリ土類金属(例えば、カルシウム及びマグネシウム)の他に製薬業界で公知の酸が含まれる。特に、医薬的に許容され得る塩の例は、生理学的に許容され得るアニオンを形成する酸と形成される有機酸付加塩、例えばトシル酸塩、メタンスルホン酸塩、酢酸塩、クエン酸塩、マロン酸塩、酒石酸塩、コハク酸塩、安息香酸塩、アスコルビン酸塩、α−ケトグルタル酸塩及びα−グリセロリン酸塩である。適当な無機塩も形成され得、その中には硫酸塩、硝酸塩、炭酸水素酸塩、炭酸塩、塩酸塩及び臭化水素酸塩が含まれる。
【0136】
本明細書に記載されているいずれものヌクレオシドは、該ヌクレオシドの活性、バイオアベイラビリティや安定性を増加させるため、または諸特性を変化させるためにヌクレオチドプロドラッグとして投与され得る。多数のヌクレオシドプロドラッグリガンドが公知である。通常、ヌクレオシドのモノ−、ジ−またはトリホスフェートのアルキル化、アシル化または他の親油性修飾によりヌクレオチドの安定性が増加する。ホスフェート部分の1つ以上の水素原子を置換し得る置換基の例はアルキル、アリール、ステロイド、糖を含めた炭水化物、1,2−ジアシルグリセロール及びアルコールである。多くの置換基がR.Jones及びN.Bischofberger,Antiviral Research,27:1−17(1995)に記載されている。これらのいずれもが所望効果を達成するためにここに開示されているヌクレオシドと組み合わせて使用され得る。
【0137】
活性ヌクレオシドは、援用により本明細書に含まれるとする下表の参考文献に開示されているように5’−ホスホエーテル脂質または5’−エーテル脂質としても提供され得る。
【0138】
【表3】
好ましくはヌクレオシドの5’−OHでヌクレオシドに共役的に取り込まれ得る適当な親油性置換基または親油性調製物を開示している米国特許明細書の非限定例には、米国特許第5,149,794号明細書(Yatvinら,1992年9月22日)、同第5,194,654号明細書(Hostetlerら,1993年3月16日)、同第5,223,263号明細書(Hostetlerら,1993年6月29日)、同第5,256,641号明細書(Yatvinら,1993年10月26日)、同第5,411,947号明細書(Hostetlerら,1995年5月2日)、同第5,463,092号明細書(Hostetlerら,1995年10月31日)、同第5,543,389号明細書(Yatvinら,1996年8月6日)、同第5,543,390号明細書(Yatvinら,1996年8月6日)、同第5,543,391号明細書(Yatvinら,1996年8月6日)、同第5,554,728号明細書(Basavaら,1996年9月10日)が含まれ、これらはいずれも援用により本明細書に含まれるとする。本発明のヌクレオシドに結合し得る親油性置換基または親油性調製物を開示している外国特許文献には、国際特許出願公開第89/02733号パンフレット、同第90/00555号パンフレット、同第91/16920号パンフレット、同第91/18914号パンフレット、同第93/00910号パンフレット、同第94/26273号パンフレット、同第96/15132号パンフレット、欧州特許出願公開第0 350 287号明細書、欧州特許出願第93917054.4号明細書及び国際特許出願公開第91/19721号パンフレットが含まれる。
【0139】
本発明の医薬組成物を製造するためには、治療有効量の1つ以上の本発明の化合物を剤形を作成するための慣用の製薬調合技術に従って医薬的に許容され得る担体と混合することが好ましい。担体は投与、例えば静脈内または非経口投与するのに望ましい剤形に応じて各種形態をとり得る。経口剤形の医薬組成物を製造するには、通常の医薬用媒体を使用し得る。よって、懸濁液、エリキシル剤及び溶液のような液体経口組成物の場合には、水、グリコール、オイル、アルコール、矯臭剤、保存剤、着色剤等を含む適当な担体及び添加剤を使用し得る。散剤、錠剤、カプセル剤のような固体経口組成物、坐剤のような固体組成物の場合、スターチ、糖担体(例えば、デキストロース、マンニトール、ラクトース及び関連担体)、希釈剤、顆粒化剤、滑沢剤、結合剤、崩壊剤等を含む適当な担体及び添加剤を使用し得る。所望により、徐放性とするために錠剤またはカプセル剤に一般的技術により腸陽性コーティングを施してもよい。前記剤形を使用すると患者における化合物のバイオアベィラビリティが有意に影響され得る。
【0140】
非経口処方物の場合、担体は通常滅菌水または塩化ナトリウム水溶液を含む、分散を助けるものを含めた他の成分も配合され得る。滅菌水を使用し、滅菌物として維持しようとするときには、組成物も担体も滅菌しなければならない。注射用懸濁液も調製可能であるが、この場合適当な液体担体、懸濁剤等が使用され得る。
【0141】
(ウイルス抗原を標的とするリポソームを含めた)リポソーム懸濁液も医薬的に許容され得る担体を作成するために慣用の方法により調製され得る。これは本発明のヌクレオシド化合物の遊離ヌクレオシド、アシルヌクレオシドまたはホスフェートエステルプロドラッグ形態をデリバリーするために適当であり得る。
【0142】
加えて、本発明の化合物は1つ以上の抗ウイルス剤、抗−HIV剤、抗−HBV剤、抗−HCV剤、抗肝炎剤またはインターフェロン、抗癌剤、抗細菌剤、または他の化合物と一緒にまたは交互に投与され得る。好ましい化合物には、α−インターフェロン及びリバビリンが含まれる。本発明の特定化合物は他の化合物の代謝、異化または不活化を減らすことにより本発明の特定化合物の生物学的活性を高めるために有効であり得、よって所期目的のために同時投与され得る。
【0143】
追加実施態様では、場合により本明細書に開示されている医薬的に許容され得る担体または希釈剤中に含まれる医薬的有効量のゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグを、場合により1つ以上の他の抗ウイルス剤と一緒または交互に哺乳動物に投与することを含む、ウイルス関連疾患を患っている哺乳動物の治療または予防方法を提供する。好ましい実施態様において、哺乳動物はヒトである。
【0144】
特に、本発明は、肝炎ウイルス属(HCV)、ペスチウイルス属(BVDV、CSFV、BDV)またはフラビウイルス属(デング熱ウイルス、西ナイルウイルスを含めた日本脳炎ウイルス及び黄熱ウイルス)のすべてのメンバーを含めたフラビウイルス科ウイルス感染を治療または予防するための方法及びそのための使用を包含する。
【0145】
本発明を以下に更に説明する。本明細書中の実施例は本発明の理解を助けるために提示されている。実施例は請求の範囲に記載されている本発明を限定するものではなく、限定するものと解釈されるべきでない。
【0146】
フラビウイルス科ウイルス感染を治療するための治療法
抗ウイルス剤で長期間治療した後にウイルスの薬剤耐性変異株が出現し得ることは認められている。最も典型的には、薬剤耐性はウイルス複製サイクルで使用される酵素(最も典型的には、HCVの場合RNA依存性RNAポリメラーゼ)をコードする遺伝子の変異により生じる。ウイルス感染に対する薬物の効果は化合物を主薬物により生ずる変異とは異なる変異を誘発する第2及び多分第3抗ウイルス化合物と一緒または交互に投与することにより延長、増強または回復させることができることが立証されている。また、薬物の薬物動態、体内分布または他のパラメーターは前記した併用または交互治療により変化させることができる。通常、併用治療はウイルスに対して複数の同時ストレスを誘発するので、通常併用治療が交互治療よりも好ましい。
【0147】
フラビウイルス科ウイルス、特にC型肝炎ウイルスに対して活性であると同定され、よってゲムシタビン、その塩、プロドラッグまたは誘導体と一緒または交互に使用され得る薬物の例を下記する:
(1) インターフェロン及び/またはリバビリン。
【0148】
(2) α−ケトアミド及びヒドラジノウレアを含めた基質ベースのNS3プロテアーゼ阻害剤(Attwoodら,「抗ウイルスペプチド誘導体(Antiviral peptide derivatives)」,国際特許出願公開第98/22496号パンフレット(1998);Attwoodら,Antiviral Chemistry and Chemotherapy,10:259−273(1999);Attwoodら,「抗ウイルス剤としてのアミノ酸誘導体の製造及び使用(Preparation and use of amino acid derivatives as anti-viral agents)」,独国特許出願公開第19914474号明細書;Tungら,「セリンプロテアーゼ、特にC型肝炎ウイルスNS3プロテアーゼの阻害剤(Inhibitors of serine proteases, particularly hepatitis C virus NS3 protease)」,国際特許出願公開第98/17679号パンフレット、及びボロン酸またはホスホネートのような求電子を停止させる阻害剤(Llinas−Brunetら,「C型肝炎阻害剤ペプチドアナログ(Hepatitis C inhibitor peptide analogues)」,国際特許出願公開第99/07734号パンフレット)。
【0149】
(3) 2,4,6−トリヒドロキシ−3−ニトロ−ベンズアミド誘導体のような非基質ベースの阻害剤(K.Sudoら,Biochemical and Biophysical Research Communications,238:643−647(1997);K.Sudoら,Antiviral Chemistry and Chemotherapy,9:186(1998))。例えば、アミド上を14炭素鎖で置換したRD3−4082及びp−フェノキシフェニル基をプロセッシングするRD3−4078。
【0150】
(4) NS3/4A融合タンパク質及びNS5A/5B基質を用いる逆相HPLCアッセイで相対阻害を示すチアゾリジン誘導体(K.Sudoら,Antiviral Research,32:9−18(1996))、特に長アルキル鎖で置換された融合シンナモイル部分を有する化合物RD−1−6250、RD4 6205及びRD4 6193。
【0151】
(5) N.Kakiuchiら,J.EBS Letters,421:217−220;N.Takeshitaら,Analytical Biochemistry,247:242−246(1997)が同定したチアゾリジン及びベンズアニリド。
【0152】
(6) SDS−PAGE及びオートラジオグラフィーアッセイにおいてプロテアーゼに対して活性を有する、フェナン−スレンキノン。ストレプトミセス種の発酵培養ブロスから単離したSch 68631(M.Chuら,Tetrahedron Letters,37:7229−7232(1996))、真菌Penicillium griscofuluumから単離したSch 351633。これらはシンチレーションプロキシミティアッセイにおいて活性を示す(M.Chuら,Bioorganic and Medicinal Chemistry Letters,9:1949−1952))。
【0153】
(7) ヒルから単離した高分子elgin cをベースとする選択的N3阻害剤(M.A.Qasimら,Biochemistry,36:1598−1607(1997))。
【0154】
(8) ヘリカーゼ阻害剤(G.D.Dianaら,「C型肝炎治療のための化合物、組成物及び方法(Compounds, compositions and methods for treatment of hepatitis C)」,米国特許第5,633,358号明細書;G.D.Dianaら,「ピペリジン誘導体、その医薬組成物及びC型肝炎治療におけるその使用(Piperidine derivatives, pharmaceutical compositions thereof and their use in the treatment of hepatitis C)」,国際特許出願公開第97/36554号パンフレット)。
【0155】
(9) ポリメラーゼ阻害剤、例えばヌクレオチドアナログのグリオトキシン(R.Ferrariら,Jouranl of Virology,73:1649−1654(1999))及び天然産物のセルレニン(V.Lohmannら,Virology,249:108−118(1998))。
【0156】
(10) ウイルスの5’−非コード領域(NCR)中の配列ストレッチに相補的なアンチセンスホスホロチオエートオリゴデオキシヌクレオチド(S−ODN)(M.Altら,Hepatology,22:707−717(1995))、またはNCRの3’末端を含むヌクレオチド326−348及びHCV RNAのコアコード領域に位置するヌクレオチド371−388(M.Alaら,Archives of Virology,142:589−599(1997);U.Galderisi,Journal of Cellular Physiology,181:251−257(1999))。
【0157】
(11) IRES依存性翻訳の阻害剤(N.Ikedaら,「C型肝炎の予防及び治療剤(Agent for the prevention and treatment of hepatitis C)」,特開平08/268890号公報:Y.Kaiら,「ウイルス疾患の予防及び治療(Prevention and treatment of viral diseases)」,特開平10/101591号公報)。
【0158】
(12) ヌクレアーゼ耐性リボザイム(D.J.Maccjakら,Hepatology,30,abstract 995(1999))。
【0159】
(13) ヌクレオシド類縁体もフラビウイルス科ウイルス感染の治療のために開発された。
【0160】
(14) Idenix Pharmaceuticals,Ltd.の国際特許出願公開第01/90121号パンフレット(2001年5月23日出願)及び同第01/92282号パンフレット(2001年5月26日出願)は分枝鎖ヌクレオシド及びそのHCV、フラビウイルス及びペスチウイルスの治療における使用を開示している。Idenixパンフレットには、場合により医薬的に許容され得る担体中に含まれる有効量の生物学的に活性な1’,2’,3’または4’−分枝鎖β−Dまたはβ−L−ヌクレオシド、またはその医薬的に許容され得る塩もしくはプロドラッグを単独または組み合わせて投与することを含むヒト及び他の宿主動物におけるC型肝炎(及び、フラビウイルス及びペスチウイルス)感染の治療方法が開示されている。
【0161】
(15) Indenix Pharmaceuticals,Ltd.の国際特許出願公開第01/96353号パンフレット(2001年6月15日出願)はHBVの治療のための2’−デオキシ−β−L−ヌクレオシドの3’−プロドラッグを開示している。Beauchampの米国特許第4,957,924号明細書はアシクロビルの各種治療用エステルを開示している。
【0162】
(16) C型肝炎ウイルスを治療するために特定のヌクレオシド類縁体を使用することを開示している他の特許文献には、BioChem Pharma,Inc.(現Shire Biochem,Inc.)の国際特許出願公開第01/32153号パンフレット(PCT/CA00/01316,2000年11月3日出願)及び同第01/60315号パンフレット(PCT/CA01/00197,2001年2月19日出願);Merck & Co.,Inc.の国際特許出願公開第02/057425号パンフレット(PCT/US02/01531,2002年1月18日出願)及び同第02/057287号パンフレット(PCT/US02/03086,2002年1月18日出願);Rocheの国際特許出願公開第02/18404号パンフレット(PCT/EP01/09633,2001年8月21日出願);Pharmassetの国際特許出願公開第01/79246号パンフレット(2001年4月13日出願)及び同第02/32920号パンフレット(2001年10月18日出願)が含まれる。
【0163】
(17) 1−アミノ−アルキルシクロヘキサン(Goldらの米国特許第6,034,134号明細書)、アルキル脂質(Chojkierらの米国特許第5,922,757号明細書)、ビタミンE及び他の抗酸化剤(Chojkierらの米国特許第5,922,757号明細書)、スクアレン、アマンタジン、胆汁酸(Ozekiらの米国特許第5,846,964号明細書)、N−(ホスホノアセチル)−L−アスパラギン酸(Dianaらの米国特許第5,830,905号明細書)、ベンゼンジカルボキサミド(Dianaらの米国特許第5,633,388号明細書)、ポリアデニル酸誘導体(Wangらの米国特許第5,496,546号明細書)、2’,3’−ジデオキシイノシン(Yarchoanらの米国特許第5,026,687号明細書)及びベンズイミダゾール(Colacinoらの米国特許第5,891,874号明細書)を含めた他の各種化合物。
【0164】
(18) C型肝炎ウイルスの治療のために現在臨床開発されている他の化合物には、Schering−Ploughのインターロイキン−10、InterneuronのIP−50、VertexのMerimebodib VX−497、Endo Labs SolvayのAMANTADINE(Symmetrel)、RPIのHEPTAZYME、Idun Pharm.のIDN−6556、XTLのXTL−002、ChironのHCV/MF59、NABIのCIVACIR、ICNのLEVOVIRIN、ICNのVIRAMIDINE、Sci CloneのZADAXIN(チロシンα−1)、MaximのCEPLENE(ヒスタミン二塩酸塩)、Vertex/Eli LillyのVX950/LY570310、Isis Pharmaceutical/ElanのISIS 14803、Idun PharmaceuticalsのIDN−6556及びAKROS PharmaのJTK 003が含まれる。
【0165】
(19) エモリー大学及びジョージア大学研究財団の米国特許第6,348,587号明細書は、HIV、B型肝炎、C型肝炎及び異常細胞増殖の治療のための2’−フルオロヌクレオシドの使用を開示している。
【0166】
合成プロトコル
ピリミジンヌクレオシドの場合、ウリジン誘導体1(スキーム1)が出発物質であり、これを2,2’−無水誘導体2に変換し、無水ジオキサン中HFで処理する(Codingtonら,J.Org.Chem.,29:558(1964))。対応の2’−フルオロ−2’−デオキシウリジン誘導体3は40〜50%の収率で得られる。3の4位は各種方法により修飾され得る。2’−フルオロ−2’−デオキシシチジン誘導体4(R=R’=R”=H)は3からチオ化または塩素化を介する周知方法により容易に得ることができる。
【0167】
【化7】
【0168】
L−ウリジンから出発して、D−シリーズで合成したすべてのL−ヌクレオシド対応物を製造することができる。
【0169】
gem−ジフルオロヌクレオシドは、ナトリウム塩方法により2,2−ジフルオロ−1−O−アセチル−3,5−ジ−O−ベンゾイル−2−デオキシ−D−リボフラノース−2−ウロース8(スキーム2)を各種シリル化ピリミジン塩基またはプリンと縮合させることにより得ることができる。糖はReformatzky反応により2,3−O−イソプロピリデン−D−グリセラール5及びブロモジフルオロ酢酸エチル6から容易に製造され、その後保護基を酸で除去するとラクトン7が得られる。7をベンゾイル化した後DIBAL還元及びアセチル化することによりラクトンをラクトールに変換すると、8が生ずる。
【0170】
【化8】
【0171】
本発明の方法をより深く理解するために下記実施例を提示する。これらの実施例は例示にすぎず、本発明の範囲を限定することを意図するものではない。ここに記載の特定の溶媒、試薬または反応条件の代わりに同等、類似または適当な溶媒、試薬または反応条件を本発明の概念を逸脱することなく使用することができる。
【実施例1】
【0172】
フラビウイルスに対する候補化合物の抗ウイルス試験:
Huh7細胞におけるHCVレプリコン系。HCVレプリコンを有するHuh7細胞は、10% 胎仔ウシ血清、1X非必須アミノ酸、Pen−Strep−Glu(それぞれ100単位/L、100μg/L及び2.92mg/L)及び500〜1000μg/L G418を含有するDMEM培地(高グルコース、ピルビン酸塩なし)において培養し得る。抗ウイルススクリーニングアッセイは、G418を含まない同一培地において以下のように実施し得る。細胞をlog増殖相に維持するために細胞を96ウェルプレートに低密度で、例えば1000細胞/ウェルで接種する。細胞を接種した直後に試験化合物を添加し、インキュベータにおいて37℃で3〜7日間インキュベートする。その後、培地を除去し、細胞を総核酸抽出(レプリコンRNA及び宿主RNAを含む)のために作成する。次いで、レプリコンRNAをQ−RT−PCRプロトコルで増殖させ、定量し得る。レプリコンRNAの定量の観察された違いが試験化合物の抗ウイルス能力を表す1つの方法である。典型的な実験で、ネガティブコントロール及び無活性化合物環境では、かなりの量のレプリコンが産生される。このことは、両方の環境でHCV RT−PCRに対して測定された閾値サイクルが相互に近いので結論づけられ得る。前記実験で、化合物の抗ウイルス効果を表す1つの方法は試験化合物の閾値RT−PCRサイクルをネガティブコントロールの平均閾値RT−PCRサイクルで減ずることである。この値をデルタCt(ΔCtまたはDCt)と呼ぶ。3.3のΔCtはレプリコン産生の1−log低下に等しい(EC90に等しい)。2以上のΔCt値(レプリコンRNAの75%低下)でHCVレプリコンRNAレベルを低下させる化合物は抗ウイルス治療のための候補化合物である。前記候補化合物は一般式(I)を有する構造に属する。ポジティブコントロールとして、組換えインターフェロンα−2a(Roferon−A,米国ニュージャージー州に所在のHoffmann−Roche)をポジティブコントロールと一緒に用いる。
【0173】
しかしながら、このHCV ΔCt値はレプリコンコード化ウイルスRNA依存性RNAポリメラーゼの特異性パラメーターを含んでいない。典型的な環境で、化合物は宿主RNAポリメラーゼ活性及びレプリコンコード化ポリメラーゼ活性の両方を減少させ得る。従って、rRNA(または、他の宿主RNAポリメラーゼI産物)またはβ−アクチンmRNA(または、他の宿主RNAポリメラーゼII)の定量及び無薬物コントロールのRNAレベルとの比較が宿主RNAポリメラーゼに対する試験化合物の効果の相対評価である。
【0174】
HCV CΔtデータ及びrRNA ΔCtの両方を用いて、特異性パラメーターを導入し得る。このパラメーターは両方のΔCt値を相互に減ずることにより求められる。こうして、デルタ−デルタCt値(ΔΔCtまたはDDCt)が得られる。0よりも低い値はレプリコンコード化ポリメラーゼに対してより高い阻害効果があることを意味する。0以下のΔCt値は宿主rRNAレベルがレプリコンレベルよりも多く影響を受けることを意味する。概して、2以上のΔCt値は無薬物処置コントロールとは有意に異なり、認められる抗ウイルス活性を示すと見做される。しかしながら、2未満のΔCt値を有するが、限られた分子細胞毒性(rRNA ΔCT=0〜2)を有する化合物が可能性のある活性化合物である。
【0175】
別の典型的な環境で、化合物は宿主RNAポリメラーゼ活性を減少させるが、宿主DNAポリメラーゼ活性を減少させない。従って、rDNAまたはβ−アクチンDNA(または、他の宿主DNA断片)の定量及び無薬物コントロールのDNAレベルとの比較が試験化合物の細胞DNAポリメラーゼに対する阻害効果の相対評価である。
【0176】
HCV ΔCtデータ及びrDNA ΔCtを用いて、特異性パラメーターを導入し得る。このパラメーターは両方の白四角Ct値を相互に差し引くことにより求められる。こうして、ΔΔCt値が得られる。0以上の値はレプリコンコード化ポリメラーゼに対してより高い阻害効果があることを意味する。0よりも低いΔΔCt値は宿主rDNAレベルがレプリコンレベルよりも多く影響を受けることを意味する。概して、2以上のΔΔCt値は無薬物処置コントロールとは有意に異なると見做され、よって更なる評価に供される興味深い化合物である。しかしながら、2未満のΔΔCt値を有するが、限られた分子細胞毒性(rDNA ΔCT=0〜2)を有する化合物が望ましい。
【0177】
HCVレプリコンRNAレベルを特異的に減少させるが、細胞RNA及び/またはDNAレベルを僅かしか減少させない化合物が抗ウイルス治療のための候補化合物である。一般式(I)の群に属する候補化合物について(HCVを含めた)フラビウイルス科ウイルスRNAを減少させる特異的能力を評価し、有力な化合物を検出した。
【0178】
多くの研究がHCV遺伝子型1a及び1bは非タイプ1遺伝子型よりもα−インターフェロンをベースとする治療に対してより耐性であることを示している。このために、数人の医者はウイルス遺伝子型1aまたは1bに感染している患者に対して長い治療期間を処方していることがある。従って、1実施態様では、ゲムシタビンをHCV 1aまたは1bに感染している患者に対してウイルス量を減少させるのに有効な用量投与する。本発明の1実施態様では、ゲムシタビンをHCV 1aまたは1b遺伝子型を有する宿主に対してα−インターフェロンと別に投与する。更なる実施態様では、ゲムシタビンをHCV 1aまたは1b遺伝子型を有する宿主に対してα−インターフェロンと一緒に投与する。
【実施例2】
【0179】
ゲムシタビン(dFdC)の抗ウイルス活性
ゲムシタビンをDMSOに溶解し、これを自己複製HCV RNAを有するHuh7細胞の細胞モデル系の培地に0.1〜50dMの最終濃度で添加した。前記実験において化合物の抗ウイルス有効性を表す1つの方法は、試験化合物の閾値逆転写ポリメラーゼ連鎖反応(RT−PCR)サイクルをネガティブコントロールの平均閾値RT−PCRサイクルで減ずることである。この値はデルタCt(ΔCtまたはdCt)と呼ばれる。HCV ΔCtデータ及びrRNA ΔCtを利用して、特異性パラメーターが導入され得る。このパラメーターはΔCt値を相互から差し引くことにより求められる。こうして、デルタ−デルタCt値(ΔΔCtまたはddCt)が得られる。4日間インキュベートすると、レプリコンHCV RNAが用量依存的に減少した(図2)。3.3Ct値はレプリコンRNAの1−log低下に等しいので、EC90値は約70nMで達した。細胞DNAレベル(リボソームDNA)または細胞RNAレベル(リボソームRNA)の低下を更に分析したところ、宿主DNA及びRNAポリメラーゼに対するこの化合物の阻害能力を表すdCt(小)が生じた。これらの計算に基づいて、自己複製HCV RNAを有するHuh7細胞の細胞モデル系においてゲムシタビンはIC50(0.240μM)よりも低い濃度でHCV RNAレベルを有意に低下させた(EC50=0.040μM)。興味深いことに、不活性代謝物のdFdU(7.0μM)はHCVレリプリコン系においてdFdCに類似の活性を示した[dCT HCV=6.39、dCt rRNA=1.96及びddCt:4.42、(EC50及びIC50データは入手できず)]。
【実施例3】
【0180】
ヒトに1回治療後のゲムシタビンの抗ウイルス活性
HCVに感染した多病巣性HCC、肝硬変及び虚血性肝炎を呈している男性患者にオキサリプラチンと一緒にゲムシタビンHCl 1200mgを1000分間かけて投与した。耐性は許容でき、よって翌日患者に再びゲムシタビン約700mmgを投与した。第2回投与前のベースラインのウイルス量は6.49logコピー/mlであった。心臓に問題が生じたので、約700mgを投与後ゲムシタビンの注入を停止した。第2回投与から8時間後のHCV RNA測定値は4.04logコピー/mlであった。これは8時間で約2.5logの低下を示した。
【図面の簡単な説明】
【0181】
【図1】ゲムシタビン及びDNA修復の自己増強作用を示す。
【図2】ゲムシタビンを用いる治療に基づくレプリコンHCV RNAの用量依存的減少を示すグラフである(黒菱形:HCV RNAに対するΔCt)。このウイルス減少を細胞DNAレベル(リボソームDNA)または細胞RNAレベル(リボソームRNA)の減少と比較して、治療指数ΔΔCt値(黒三角:HCV−rDNA ΔΔCt、太字X:HCV−rRNA ΔΔCt)を求めた。
【技術分野】
【0001】
本発明は、ゲムシタビン、またはその医薬的に許容され得る塩、プロドラッグもしくは誘導体を用いるフラビウイルスまたはペスチウイルス、特にC型肝炎ウイルス(HCV)を治療するための方法及び投薬計画である。
【背景技術】
【0002】
ゲムザール(登録商標)(ゲムシタビンHCl)は白血病及び各種固形腫瘍(例えば、膵臓癌、非小細胞肺癌、卵巣癌、乳癌、中皮腫等)に対して抗腫瘍活性を有するピリミジン代謝拮抗物質である。ゲムシタビンは式β−D−2’,2’−ジフルオロシチジン(下記構造式参照)を有するヌクレオシド類縁体である。ゲムシタビンは元々抗ウイルス効果について研究されたが、その後抗悪性腫瘍剤として開発された(D.C.Delong,L.W.Hertel及びJ.Tang,「2’,2’−ジフルオロデオキシシチジンの抗ウイルス活性(Antiviral activity of 2',2' Difluorodeoxycytidine)」,American Society of Microbiology(1986))。ゲムシタビンは食品医薬品局(FDA)により次の適応症に認可されている:(1)手術不能な局所的に進行した(IIIA期またはIIIB期)または転移(IV期)非小細胞肺癌(NSCLC)を患っている患者に対する第一選択治療としてシスプラチンと併用して、(2)膵臓の局所的に進行した(切除不能なII期またはIII期)または転移(IV期)腺癌を患っている患者に対する第一選択治療として、及び(3)5−フルオロウラシル(5−FU)の治療を受けた患者における膵臓癌に対する第二選択治療として。
【0003】
【化4】
【0004】
ゲムシタビン(dFdC)はDNA合成(S期)を経ている細胞を主に標的とする細胞周期特異的物質である。ゲムシタビンは細胞内で律速酵素のデオキシシチジンキナーゼ(dCK)によりそのモノホスフェート形態(dFdCMP)に代謝される(V.Heinemannら,「2’,2’−ジフルオロデオキシシチジン及び1−β−D−アラビノフラノシルシトシンの細胞薬物動態及び毒性の比較(Comparison of the Cellular Pharmacokinetics and Toxicity of 2',2'-Difluorodeoxycytidine and 1-Beta-D-Arabinofuranosylcytosine)」,Cancer Res.,48(14):4024−31(1988))。その後他のヌクレオシドキナーゼによりリン酸化されると、活性な代謝物dFdCDPdF及びdFdCTPが形成される。ゲムシタビンの細胞毒性はジホスフェート及びトリホスフェート代謝物による作用の組合せに起因し、前記代謝物によりゲムシタビンの致死作用が高められる。これらの作用を図1に要約する。まず、dFdCDPはリボヌクレオチドレダクターゼを阻害し(経路1)、これによりDNA複製に必要な細胞デオキシヌクレオチド(例えば、デオキシシチジントリホスフェート,dCTP)の濃度が低下する(図1;ゲムシタビン及びDNA修復の自己増強作用)。リボヌクレオチドレダクターゼの阻害により細胞dCTP濃度が低くなると、ゲムシタビン誘導致死率にとって重要な事象であるDNAへのdFdCTPアナログの取り込みが助けられる(経路2)(P.Huang及びW.Plunkett,「フルダラビン及びゲムシタビン誘導アポトーシス:アナログのDNAへの取り込みは重要な事象である(Fludarabine- and Gemcitabine-Induced Apoptosis: Incorporation Of Analogs Into DNA Is A Critical Event)」,Cancer Chemother.Pharmacol.,36(3):181−8(1995);P.Huang及びW.Plunkett,「ゲムシタビンによるアポトーシスの誘導(Indudction Of Apoptosis By Gemcitabine)」,Semin.Oncol.,22(4 補遺11):19−25(1995))。高dCTPレベルは律速酵素dCKを阻害するので、細胞dCTPレベルが低下するとゲムシタビンリン酸化速度も増加する(経路3)。dCKに対する阻害作用と対照的に、dCTPはゲムシタビンヌクレオチドの細胞からの消失のための律速酵素であるdCMPデアミナーゼ活性のために必要なコファクターである(経路4)。細胞毒性代謝物dFdCTPは直接dCMPデアミナーゼを阻害する(経路5)(Y.Z.Xu及びW.Plunkett,「2’,2’−ジフルオロデオキシシチジンの完全ヒト白血病細胞作用におけるデオキシシチジレートデアミナーゼの調節(Modulation Of Deoxycytidylate Deaminase In Intact Human Leukemia Cells Action of 2',2'-difluorodeoxycytidine)」,Biochem.Pharmacol.,44(9):1819−27(1992))。最後に、高細胞内濃度でFdCTPはCTPシンターゼを阻害し(経路6)、よってCTPの合成が阻止され、その結果dCTPの合成も阻止される(V.Heinemannら,「ゲムシタビン:細胞内ヌクレオチド及びデオキシヌクレオチド代謝の調節剤(Gemcitabine: A Modulator of Intracellular Nucleotide And Deoxynucleotide Metabolism)」,Semin.Oncol.,22(4 補遺11):11−8(1995))。
【0005】
ゲムシタビンは、dCKによるdFdCMPからのモノホスフェートへのリン酸化に対する良好な基質であり、デオキシシチジン(チャイニーズハムスター卵巣(CHO)細胞由来の部分的に精製した酵素を用いて測定して、Km=1.6μmol/L)と類似の基質効率(Vmax/Km)で3.6μmol/LのKmを示す(V.Heinemannら,「2’,2’−ジフルオロデオキシシチジン及び1−β−D−アラビノフラノシルシトシンの細胞薬物動態及び毒性の比較(Comparison of the Cellular Pharmacokinetics and Toxicity of 2',2'-Difluorodeoxycytidine and 1-Beta-D-Arabinofuranosylcytosine)」,Cancer Res.,48(14):4024−31(1988))。ゲムシタビンのリン酸化はその生物学的活性にとって必須であり、dCKを欠く細胞はゲムシタビンにより影響されない。DNA、RNA及びタンパク質合成の放射活性前駆体を用いる研究で、ゲムシタビンの効果は主にDNAに向けられることが立証された(W.Plunkettら,「ゲムシタビン:前臨床薬理学及び作用機序(Gemcitabine: Preclinical Pharmacology And Mechanisms Of Action)」,Semin.Oncol.,23(5 補遺10):3−15(1996))。DNA合成のモデル系で、トリホスフェートdFdCTPがヒトDNAポリメラーゼα及びεにより成長DNAプライマー鎖に組み込まれ、各ポリメラーゼが正常ヌクレオチド(dCTP)よりも20倍優先的であることを示すことが確認された(P.Huangら,「DNA合成に対する2’,2’−ジフルオロデオキシシチジンの作用(Action of 2',2'-Difluorodeoxycytidine on DNA Synthesis)」,Cancer Res.,51(22):6110−7(1991))。ユニークなことに、ゲムシタビンの取り込みの後1つ以上のヌクレオチドを付加した後にDNAポリメラーゼが阻害される。終わりから2番目の位置に置くと、3’→5’プロフリーディングエキソヌクレアーゼによるdFdCMPによる切除はdCMPの切除よりも非常にゆっくり進行する。“仮面連鎖停止”として記載されているこの現象により、ゲムシタビンのDNA複製及び修復を阻害する能力が改良され、ゲムシタビンのDNA障害剤(例えば、シスプラチン)との相乗作用のメカニズムが与えられる。
【0006】
ゲムシタビンは、臨床使用中に脱アミノ化生成物の2’,2’−ジフルオロデオキシウリジン(dFdU)への生体内変化を介する迅速な代謝クリアランスに関与する酵素である細胞内シチジンデアミナーゼ(Km=96μM)に対する良好な基質である(D.Y.Bouffard,J.Laliberte及びR.L.Momparler,「2’,2’−ジフルオロデオキシシチジン(ゲムシタビン)と精製ヒトデオキシシチジンキナーゼ及びシチジンデアミナーゼに関するカイネティック研究(Kinetic Studies On 2',2'-Difluorodeoxycytidine (Gemcitabine) With Purified Human Deoxycytidine Kinase And Cytidine Deaminase)」,Biochem.Pharmacol.,45(9):1857−91(1993))。ゲムシタビンは血液、肝臓、腎臓及び他の組織中で急速に脱アミノ化される。ゲムシタビン処分が放射標識した薬物100mg/m2を1回30分間注入により投与した5人の被験者において評価された。ゲムシタビン(<10%)及び不活性代謝物dFdUは排泄用量の99%を占めた。代謝物dFdUは血漿中でも検出され、ゲムシタビン血漿タンパク質結合は無視できた。
【0007】
ゲムシタビンの薬物動態をいろいろな固形腫瘍を患っている353人の患者(2/3が男性)で研究した。薬物動態パラメータは、短い(<70分)及び長い(70〜285分)注入時間で週1回投与し、周期的に数週間休薬して異なる期間治療した患者からのデータを用いて調べた。投与されたゲムシタビン総用量は500〜3600mg/m2であった。ゲムシタビン薬物動態は直線的であり、2−区画モデルにより記載されている。消失は腎排泄に依存し、クリアランスは年齢及び性の影響を受けた。1回及び複数回投与研究の集団薬物動態分析から、ゲムシタビンの分布容量は注入時間及び性により大きく影響されたことが分かった。注入時間が短いときのゲムシタビン半減期は32〜92分であり、注入時間が長いときのゲムシタビン半減期は245〜638分であった。これらのデータは注入時間を長くすると分布容量が多くなることを反映した。分布容量は注入時間が短い(<70分)ときには50L/m2であり、ゲムシタビンが組織中に広く分布しないことを示している。反対に、注入時間が長いときの分布容量は370L/m2に増加し、これはゲムシタビンが組織区画内でゆっくり平衡化することを反映している。代謝物は週投1回の投与で蓄積されなかったが、その消失は腎排泄に依存し、dFdUレベルは腎不全により影響され得る。
【0008】
ゲムシタビン処分に対する有意な腎または肝不全の影響は今まで評価されていない。活性代謝物dFdCTPは末梢血単核細胞から抽出され得、単核細胞からのdFdCTPの終相半減期は1.7〜19.4時間である。
【0009】
重要なことに、最大耐量(MTD)は注入スケジュール及び注入頻度に大きく依存する(E.Bovenら,「実験的ヒト癌における抗腫瘍効果に対するゲムシタビンのスケジュール及び用量の影響(The Influence Of The Schedule And The Dose Of Gemcitabine On The Antitumour Efficacy In Experimental Human Cancer)」,Br.J.Cancer,68(1):52−6(1993))。60分を越える注入時間の延長及び週1回以上の投与回数はゲムシタビン関連毒性を高めることが判明した。典型的には、骨髄抑制は白血病、血小板減少症及び貧血を発現する用量限定的毒性である。血液学的毒性のために用量を頻繁に調節する必要があるので、患者は治療中骨髄抑制についてモニターされなければならない。ゲムシタビンに関連する他の毒性には、口内炎、悪心、嘔吐、発熱、発疹、軽い不規則性機能亢進(parasthesias)、軽い脱毛、インフルエンザ様症状(すなわち、発熱、悪寒、筋肉痛、咳及び頭痛)、呼吸困難、浮腫、軽度の蛋白尿及び血尿、1つまたは両方の血清トランスアミナーゼの一過性上昇及び下痢が含まれる。2つの臨床治験において、局所的に進行した膵臓癌または転移性膵臓癌患者でゲムシタビンの効果を評価した。第1の治験では、過去に化学療法を受けたことのない患者でゲムシタビンと5−FUを比較し、第2の治験では過去に5−FUまたは5−FU含有投薬を用いる治療を受けたことのある患者を評価した。いずれの治験でも、ゲムシタビンは1000mg/m2の用量で30分間注入により週1回連続7週間(または、投薬中止が必要となる毒性が生ずるまで)投与した後1週間治療を休止した。その後のサイクルは週1回連続3週間注入した後、1週間休止とした。この研究での主たる効果パラメーターは、鎮痛剤消費量、痛み強度、パフォーマンス状態及び体重変化に基づく改良により定義され且つ測定される臨床的に有利な応答に基づいていた。第1の治験では、局所的に進行した膵臓癌または転移性膵臓癌患者に対してゲムシタビン及び5−FUを多施設において予測的一重盲検でランダム比較した(H.A.Burrisら,「進行膵臓癌患者に対する第一選択治療としてのゲムシタビンを用いる生存率及び臨床効果の改良:無作為化治験(Improvements in Survival And Clinical Benefit With Gemcitabine As First-Line Therapy For Patients With Advanced Pancreas Cancer: A Randomized Trial)」,J.Clin.Oncol.,15(6):2403−13(1997))。各治療群で評価した63患者で、ゲムシタビンは5−FUと比較して臨床効果応答、生存率及び病気進行までの時間の点で統計的に有意な向上を示した。第2の治験では、過去に5−FUまたは5−FU含有投薬の治療を受けたことのある63患者に対してゲムシタビンを多施設においてオープンラベル試験した(M.L.Rothenbergら,「5−FU耐性膵臓癌患者におけるゲムシタビンの第II相治験(A Phase II Trial Of Gemcitabine In Patients With 5-FU-Refractory Pancreas Cancer)」,Ann.Oncol.,7(4):347−53(1996))。前記研究で、平均3.9ヶ月の生存率で27%の臨床効果応答率を示した。
【0010】
2つの無作為化研究(657患者)のデーターから、局所的に進行したまたは転移NSLC患者の第一選択治療のためにゲムシタビンとシスプラチンを併用することが裏付けられている。第1の研究でゲムシタビン+シスプラチンとシスプラチン単独を比較し、第2の研究でゲムシタピン+シスプラチンとエトポシド+シスプラチンを比較した。第1の研究では全部で522被験者を対象とした(P.L.Mitchell,「クオリティオブライフ及びシスプラチン−ゲムシタビン化学療法(Quality Of Life And Cisplatin-Gemcitabine Chemotherapy)」,J.Clin.Oncol.,18(14):2791−2(2000))。ゲムシタビン(1000mg/m2)を28日周期の1日目、8日目及び15日目に投与し、シスプラチン(100mg/m2)は各周期の1日目に投与した。ゲムシタビン+シスプラチン治療群での平均生存時間及び病気進行までの平均時間はシスプラチン単独群に比して有意に長かった。客観的応答率はゲムシタビン+シスプラチン治療群で26%であり、シスプラチン単独では10%であった。第2の多施設研究では、IIIB期またはIV期のNSCLCの135患者に対して21日周期の1日目及び8日目にゲムシタビン(1250mg/m2)と1日目にシスプラチン(100mg/m2)、または21日周期の1日目、2日目及び3日目にエトポシド(100mg/m2)静注と1日目にシスプラチン(100mg/m2)を用いて治療した(F.Cardenalら,「局所的に進行したまたは転移性非小細胞肺癌の治療におけるゲムシタビン−シスプラチン対エトポシド−シスプラチンの無作為化III相研究(Randomized Phase III Study Of Gemcitabine-Cisplatin Versus Etoposide-Cisplatin In The Treatment Of Locally Advanced Or Metastatic Non-Small-Cell Lung Cancer)」,J.Clin.Oncol.,17(1):12−18(1999))。2つの治療群で生存率に有意な差はなかった。しかしながら、ゲムシタビン+シスプラチン治療群の病気進行までの平均時間及び客観的応答率がエトポシド+シスプラチン治療群に比して有意に長かった。
【0011】
1997年以来、ゲムシタビン治療に関連する急性呼吸窮迫症候群(ARDS)が数例報告されている。これらの症例は有意な罹患率及び死亡率を伴っている(Sabria−Triasら,Rev.Mal.Respir.,19:645−7(2002);Guptaら,Am.J.Clin.Oncol.,25(1):96−100(2002))。ゲムシタビンの肺毒性はゲムシタビン誘導全身性毛細管漏出症候群(SCLS)の検出に関連している。ゲムシタビン誘導SCLSは急速に進行する浮腫、体重増加、低血圧、血液濃縮及び低蛋白血症により特徴づけられ、血管内から間質スペースへの血漿の急速管外遊出を伴う突然の可逆的毛細管透過性亢進に起因する高い死亡率を有するまれな疾患である。最近の証拠で、ゲムシタビンSCLSはゲムシタビンの肺毒性に関する病原メカニズムであることが示唆されている(DePasoら,Ann.Oncol.,12(11):1651−2(2001))。更に、ゲムシタビンの効果及び安全性は用量よりもスケジュールにより強く依存していることも報告されている(Vermorkenら,Br.J.Cancer,76(11):1489−93(1997))。
【0012】
ゲムシタビンは抗癌剤として開発されたが、抗ウイルス剤としてのゲムシタビンはほとんど熱心に研究されていなかった。なぜならば、(i)ゲムシタビンを抗腫瘍剤として熟知している人々は、ゲムシタビンは非常に有毒なので、典型的には、週1回3〜4週間投与した後「休止週間」という投薬計画に従ってしか通常投与されないことを知っているから(下表1参照)、及び(ii)標準の抗ウイルス治療はヌクレオシド類縁体を無期限に、多分患者の一生の間毎日投与することからなるからである。
【0013】
【表1】
【0014】
【表2】
【0015】
表2の精査から、抗ウイルス剤治療はウイルス量の1〜2log低下を維持するために長期間にわたり毎日投与する必要があることが示される。通常、ウイルス学者は、抗ウイルス剤の治療を中止した(または、時々しか投与しなかった)場合にウイルスが消滅されていなかったならば、ウイルス量は急速にリバウンドし、持続的治療効果が得られないことを認めている。
【0016】
1986年にLilly Research LaboratoriesのDelongらは、
「2’,2’−ジフルオロ−2’−デオキシリボースを含有するヌクレオシド群を合成してその抗ウイルス活性を試験できた。前形成単層において毒性を呈することなくRNA及びDNAウイルスの両方に対して非常に高いインビトロ活性を有しているシチジンアナログが特に興味深い。この化合物は、チミジンキナーゼネガティブであり、改変DNAポリメラーゼを有するFMAU及びアシクログアノシンに対して耐性のHSV−1変異体をも阻害した。毒性は培養中の急速に増殖する細胞で観察された。前記化合物の抗ウイルス効果について各種動物モデルで試験した。化合物は動物の急性ウイルス感染においてウイルス増加を抑制したが、我々は毒性とウイルス活性を区別することはできなかった。しかしながら、我々は、マウスのフレンド白血病ウイルス感染において投薬スケジュールを変えることにより毒性と区別できる非常に高い活性を観察した。脾臓肥大及び多赤芽球症は体重が正常に増加する条件で90%抑制できた。5日毎に治療を要する投薬スケジュールが可能であった。経口及びIP経路で活性が観察された。感染のために脾臓が肥大したマウスの脾臓を治療により縮小できたことを示す研究を実施した。」
という抄録を発表した。
【0017】
強調は、追記した。米国微生物学会の年次総会の抄録(1986;Abstract No.T−56)。著者はウイルス感染(フレンド白血病ウイルス)の1症例で活性を毒性と区別できたと報告したが、これは明らかに毒性と活性を区別できないことを示す報告パターンの孤立した例外である。
【0018】
米国特許第5,015,743号明細書は、ウイルス疾患の治療のためのゲムシタビンを含めた2,2−ジフルオロ−2−デスオキシ炭水化物ヌクレオシドの属を開示している。この特許文献は、「この発明の抗ウイルス性ヌクレオシドはウイルス感染の治療のためにこの病気の治療における通常の方法で使用される」と教示している。実際、ゲムシタビンは一般的な抗ウイルス治療に従って毎日無期限に投与することはできないことは現在公知である。この特許文献はインビトロ生物学的活性の実施例を1つ含んでいるが、この“テスト1”は試験した化合物を明確に同定していない。毒性を調べるインビボデータは提示されていない。
【0019】
Hoffmann−LaRoche社が出願した国際特許出願公開第02/18404号パンフレット及び米国特許出願公開第2003/0008841号明細書は、C型肝炎を治療するための特定ヌクレオシド誘導体を開示している。ゲムシタビンはこの出願の表1、実施例243に示されている化合物243である。投薬に関して、Rocheの明細書は、
「C型肝炎ウイルス感染の治療のために必要な式Iを有する化合物の量は病気の重篤度、レシピエントの種類、性別及び体重を含めた多数の要因に依存し、最終的には担当医の裁量にまかされる。しかしながら、通常適当な有効量は0.05〜100mg/レシピエントの体重kg/日の範囲であり、好ましくは0.1〜50mg/体重kg/日の範囲、最も好ましくは0.5〜20mg/体重kg/日の範囲である。最適用量は約2〜16mg/体重kg/日である。所望用量を1日を通して適当な間隔で2回、3回、4回、5回、6回またはそれ以上に分割して投与することが好ましい。分割量は1剤形あたり例えば1〜1500mg、好ましくは5〜1000mg、最も好ましくは10〜700mgの活性成分を含む1回服用量剤形で投与され得る。」
と教示している。
【0020】
繰り返すと、ウイルス感染を治療するためにはゲムシタビンを含む上記化合物を1日数回ではないとしても、毎日用いなければならないと公衆は教示されている。毒性が公知であるので、少なくともゲムシタビンに関するこの教示は「死細胞は生存ウイルスを含まない」との古い格言の範疇に入ると見られる。しかしながら、まともな医者はウイルス感染を治療するための手段として慢性薬物中毒のために患者を殺すかまたは患者に対して重大なダメージを与えることはないであろう。従って、従来の報告に関係なく、HCVを含めたフラビウイルス感染を治療するためにゲムシタビンを使用することを誰も真剣に検討していない。
【0021】
米国特許出願公開第2002/0052317号明細書及び国際特許出願公開第02/10743号パンフレットは、インターフェロンに対する耐性を改良するためにエリスロポエチンを使用し、この療法では場合によりゲムシタビンを含むヌクレオシド類縁体の上位概念のクラスの1つを投与してもよいことを開示している。
【0022】
C型肝炎ウイルスを含むフラビウイルス科
フラビウイルスは9〜15kbのゲノムイズを有する正の一本鎖RNAウイルス群である。これらのウイルスは約45〜50nmのエンベロープウイルスである。フラビウイルス分類学の概説はウイルス分類学国際委員会から入手可能である。フラビウイルス科は3つの属からなる。
【0023】
1. フラビウイルス
この属には、デング熱ウイルス群(デング熱ウイルス、デング熱ウイルスタイプ1、デング熱ウイルスタイプ2、デング熱ウイルスタイプ3、デング熱ウイルスタイプ4)、日本脳炎ウイルス群(Alfuyウイルス、日本脳炎ウイルス、Kookaburraウイルス、Koutangoウイルス、クンジンウイルス、マレー渓谷脳炎ウイルス、セントルイス脳炎ウイルス、ストラットフォードウイルス、Usutuウイルス、西ナイルウイルス)、モドックウイルス群、リオブラボウイルス群(アポイウイルス、リオブラボウイルス、サボヤウイルス)、ヌタヤ(Ntaya)ウイルス群、マダニ伝搬脳炎ウイルス群(マダニ伝搬脳炎ウイルス)、Tyuleniyウイルス群、ウガンダSウイルス群及び黄熱ウイルス群が含まれる。これらの主要群の他に、分類されていない幾つかの追加フラビウイルスが存在する。
【0024】
2. ペスチウイルス
この属には、ウシウイルス性下痢症ウイルス−2(BVDV−2)、(BVDVを含めた)ペスチウイルスタイプ1、(豚コレラウイルスを含めた)ペスチウイルスタイプ2及び(ボーダー病ウイルスを含めた)ペスチウイルス3が含まれる。
【0025】
3. 肝炎ウイルス
この属には1つの種のみ、すなわち多くのクレード、タイプ及びサブタイプから構成されるC型肝炎ウイルス(HCV)のみが含まれる。
【0026】
HCVは1989年まで同定されておらず、それまで非A非B肝炎と称されていた。HCVはB型肝炎と合わせて世界中の肝疾患のすべての症例の75%を占めている(B.Helblingら,「未処置の慢性C型肝炎におけるインターフェロン及びアマンタジン:プラセボをコントロールとする二重盲検無作為化治験(Interferon And Amantadine In Naive Chronic Hepatitis C: A Doubleblind, Randomized, Placebo-Controlled Trial)」,Hepatology,35(2):447−54(2002))。HCV感染に関連する肝不全は米国での肝移植の主な原因である。HCV感染は通常初期段階では軽度であるので、慢性段階になるまで診断されにくい。従って、HCVはしばしば「無症候流行病」と呼ばれている。HCVは感染から肝疾患の発症までの典型型サイクルには約20年を要し、よって増えつつある感染者に対するこの病気の真の影響は多年にわたり現れない。HCVは感染者の血液と接触することにより広がる。HCV感染のリスク因子が高い人には、非合法注射薬の使用者、1992年以前に輸血または固形臓器移植を受けた人、1987年以前に凝血の問題のために血液製剤を投与された人、長期間腎臓透析を受けている患者、肝疾患の証拠を呈している人(例えば、ALTレベルが持続的に異常な人)が含まれる。
【0027】
米国では約400万人がHCVに感染しており、世界中では2億人以上が感染していると推定される(S.E.Hewitt,「C型肝炎ウイルス(HCV)感染及びHCV関連疾患の予防及びコントロールのための推奨(Recommendations for Prevention and Control of Hepatitis C Virus (HCV) infection and HCV-related Disease)」,疾病管理及び予防センター(1998))。1980年代では毎年平均230,000人が新たに感染している。1989年以降、新たに感染した患者の数は1986年までの>80%減少し、36,000人になった。これらの患者の多くは慢性的に感染しており、症状が出ないままであるのでこの感染に気づいていない場合がある。よって、HCV関連肝疾患は今世紀に直面している最大の公衆衛生脅威の1つであり得る。
【0028】
慢性肝疾患は米国の成人の死亡の主因の10位であり、毎年25,000人が死亡している。集団研究で、慢性肝疾患の40%がHCVに関連していると推定される。多くのHCV感染者は30〜49歳であるので、HCV関連の慢性肝疾患による死亡者の数は次の10〜20年に実質的に増加する恐れがある。HCV関連合併症の現在の医療費が>6億ドル/年と推定されているので、これは軽微なことでない。
【0029】
HCVはRNAウイルスであり、これは頻繁に変異することを意味する(www.epidemic.org/index2.html,「C型肝炎に関する事実(The Facts about Hepatitis C)」,1998,ダートマス大学)。一旦感染すると、HCVは宿主の体内でそれ自体の異なった遺伝的変異を生ずる。変異体はしばしばその前駆体とは異なり、免疫系は変異体を認識できない。よって、たとえ免疫系が1つの変異に対して成功しても、変異株はすぐに支配的になり、優勢株となる。このことが、HCV感染者の>80%が慢性肝疾患に進行する理由となる。HCVは6つの主要遺伝子型及び50以上のサブタイプを有する。米国では、HCV感染患者の中で約70%が遺伝子型1を有し、15%が遺伝子型2を有し、10%が遺伝子型3を有する(J.G.McHutchisonら,「慢性C型肝炎の初期治療としてのインターフェロン Alfa−2b単独とリバビリンとの併用(Interferon Alfa-2b Alone Or In Combination With Ribavirin As Initial Treatment For Chronic Hepatitis C)」,Hepatitis Interventional Therapy Group,N.Engl.J.Med.,339(21):1485−92(1998))。肝硬変への進行のリスクを有する慢性HCV感染患者に対して抗ウイルス治療が推奨されている(S.K.Herrine,「慢性C型肝炎ウイルス感染患者に対するアプローチ(Approach To The Patient With Chronic Hepatitis C Virus Infection)」,Ann.Intern.Med.,136(10):747−57(2002))。上記した患者には、ALTレベルが持続的に高く、HCV RNAが検出可能であり、肝生検で門脈または架橋状線維症を示したかまたは少なくとも軽度の炎症または壊死を示している抗−HCV抗体陽性患者が含まれる。
【0030】
HCVに対する治療は急速に変化しており、インターフェロン、リバビリンやヌクレオシド類縁体との併用治療が慢性HCV感染を有する未処置患者を治療するために米国で認可されている(S.E.Hewitt,「C型肝炎ウイルス(HCV)感染及びHCV関連疾患の予防及びコントロールのための推奨(Recommentations for Prevention and Control of Hepatitis C Virus (HCV) infection and HCV-related Disease)」,疾病管理及び予防センター(1998))。持続的応答率はインターフェロン単独で治療した患者では15〜25%でしかなかったのに対して、リバビリン+インターフェロンで治療した患者では40〜50%に達した。しかしながら、米国で最も一般的なHCV遺伝子型である遺伝子型1を有する患者での併用治療の成功率は低く、これらの患者での持続的応答率は<30%にすぎない。更に、治療に伴う副作用によりしばしば治療の用量を低下させたり、治療期間を中断させている。インターフェロン+リバビリン治療に伴ってしばしば生ずる副作用にはインフルエンザ様症候群、過敏症、うつ病、貧血、骨髄抑制及び腎不全が含まれる。リバビリンは妊娠の可能性ある女性では催奇形性であり、禁忌である。
【0031】
慢性HCV感染による公衆衛生の脅威及び現在の治療の限界のために、HCV感染を治療するための革新的治療アプローチが要望されている。
【発明の開示】
【0032】
従って、本発明の目的はフラビウイルス、特にHCV感染を治療するための新規組成物及び方法を提供することである。
【0033】
驚くべきことに、最少用量のゲムシタビン(または、本明細書に記載されているその塩、プロドラッグもしくは誘導体)によりヒト患者におけるC型肝炎のウイルス量を数日間未満、実際ある場合には1〜2日間以下で最高2logまで、またはそれ以上低下させることができることが知見された。こうしたウイルス量の急速で大きな低下は、約14日以上毎日持続的に治療した後に1〜2log安定的にしか低下できない従来の抗ウイルス治療経験と対照的である。ヒトにおけるゲムシタビン、またはその塩もしくはプロドラッグの予期しない強くユニークな抗−HCV活性により抗ウイルス薬物投与モデルの基本的変更の基礎が与えられ、前記治療のためにゲムシタビンを保存的にかつ適切に使用することが初めてできた。
【0034】
従って、第1実施態様において、本発明は、ゲムシタビン(または、本明細書に記載されているその塩、プロドラッグもしくは誘導体)を約50〜約1300mg/m2/日の範囲用量で1、2又は3日間投与した後治療を休止することを含むフラビウイルス感染、特にC型肝炎ウイルス感染を治療するための方法及び組成物を提供する。場合により、ウイルス量を経時的にモニターしてウイルスリバウンドを調べる。有意なウイルス量が再び観察されない限り治療を再開せず、観察されたときに1、2又は3日間の治療を繰り返す。この治療は患者の健康をモニターし、維持するために無期限に継続することができる。
【0035】
治療され得るフラビウイルス科ウイルスには、肝炎ウイルス属(HCV)、ペスチウイルス属(BVDV、CSFV、BDV)及びフラビウイルス属(デング熱ウイルス、西ナイルウイルスを含めた日本脳炎ウイルス群、及び黄熱ウイルス)のすべてのメンバーが含まれる。
【0036】
別の実施態様では、より重篤なフラビウイルス科感染に対してゲムシタビン(または、本明細書に記載されているその塩、プロドラッグもしくは誘導体)を約50〜約1300mg/m2/日の範囲用量で1日間〜7日間(例えば、1、2、3、4、5、6または7日間)投与した後治療を休止する。場合により、ウイルス量を経時的にモニターし、休止後のウイルスリバウンドをモニターする。有意なウイルス量が再び観察されない限り治療を再開せず、観察されたときに1〜7日間(例えば、独立して、1、2、3、4、5、6または7日間)、より好ましくは1、2又は3日間の治療は繰り返す。この治療は患者の健康をモニターし、維持するために無期限に継続することができる。
【0037】
本発明はゲムシタビン、その塩もしくはプロドラッグを用いて抗ウイルス治療を抗腫瘍投薬スケジュールを用いて実施し得ることを初めて開示している。特定実施態様では、フラビウイルス感染を治療するためにゲムシタビンに対する認可されたいずれもの抗腫瘍投薬スケジュールを使用することができる。
【0038】
幾つかの非限定的例示実施態様では、ゲムシタビンの1日用量は100〜1500mg/日、または200〜1000mg/日、特に300〜800mg/日の範囲であり得る。
【0039】
1つの例示実施態様では、1日目に患者に静注投与し、薬物投与から数時間、多分最高12時間病院に留まるよう指示する。患者を安全性及び耐性についてモニターし、HCV−RNAを調べるために投与前、投与から6時間目及び12時間目に採血する。2日目には安全性評価及びウイルス量測定のために患者に再び来院させる。場合により治療を2、3、4、5、6及び7日目にも継続する。その後、治療を休止し、患者には安全性及びウイルス量試験を追跡するために定期的に来院するように指示する。
【0040】
ゲムシタビンは消化管で急速にそのウラシル誘導体に変換されることが公知であるので、ゲムシタビンを静注の形態で投与することが好ましい。ゲムシタビンを経口投与することが好ましいならば、前記化合物をプロドラッグであって、活性に悪影響を与えることなくシトシニルアミン基が急速脱アミノ化されることを保護するプロドラッグの形態で投与することが好ましい。シトシン塩基のインビボでの半減期を延長させるための非限定的方法として、前記化合物をN−アシル化、N−アルキル化またはN−アリール化形態で投与することが挙げられる。
【0041】
プロドラッグには、本明細書に記載のヌクレオシドのエステル及びアミドを形成するためのヒドロキシまたはアミノ官能基上のアミノ酸誘導体も含まれる。例えば、アシクロビルそのものに比較して改良された水溶性を有するアシクロビルのアミノ酸エステル、特にグリシンエステル及びアラニンエステルを開示している欧州特許出願公開第99493号明細書、及びアシクロビルのアラニンエステル及びグリシンエステルに比較して経口投与後高いバイオアベイラビリティを示すα−炭素原子に隣接した側鎖分枝鎖を特徴とするアシクロビルのバリンエステルを開示している米国特許第4,957,924号明細書(Beauchamp)を参照されたい。前記アミノ酸エステルの製造方法は米国特許第4,957,924号明細書(Beauchamp)に記載されている。バリンそのものの使用に代えて、アミノ酸の機能的均等物(例えば、酸クロリドのような酸ハライドまたは酸無水物)を使用してもよい。その場合、望ましくない副作用を避けるために、アミノ保護誘導体を使用することが有利であり得る。
【0042】
本発明の1例として、HCVに感染した多病巣性HCC、肝硬変及び虚血性肝炎を呈している男性患者にゲムシタビンHCl 1200mgを1000分間かけてオキサリプラチンと一緒に投与した。耐性は許容でき、よって翌日患者には再びゲムシタビン約700mgを投与した。第2回投与前のベースラインのウイルス量は6.49logコピー/mlであった。心臓に問題が生じたので、約700mgを投与後ゲムシタビンの2回目の注入を停止した。心臓の問題は明らかにゲムシタビン治療とは関係なかった。第2回投与から8時間後のHCV RNA測定値は4.04logコピー/mlであった。8時間で約2.5logの低下を示した。
【0043】
別の実施態様では、ゲムシタビン、またはその塩、プロドラッグもしくは誘導体を1つ以上の他の抗−フラビウイルス活性剤と一緒にまたは交互に本明細書に記載の投薬計画に従って投与する。以下に詳記する他の活性剤はこの投薬計画と組み合わせてその効果を最大限とするように投与される。
【発明を実施するための最良の形態】
【0044】
驚くべきことに、最少量のゲムシタビンが数日間未満で、実際にはある場合には1〜2日間以内でヒト患者においてC型肝炎のウイルス量を最高2logまで、またはそれ以上低下させることができることが知見された。このようにウイルス量が急速に大きく低下することは、1〜2logの低下が約14日間以上毎日継続治療した後に安定的にしか達成されない従来の抗ウイルス経験と対照的である。ヒトにおけるゲムシタビンの予期せぬ強くユニークな抗−HCV活性により、抗ウイルス薬の投薬モデルの基本的変更の基礎が与えられ、初めて前記治療のために前記薬物が保存的にかつ適切に使用できた。
【0045】
従って、第1態様で、本発明は、ゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグを約50〜約1300mg/m2/日の範囲の用量で1、2又は3日間投与した後治療を休止することを含むフラビウイルス感染、特にC型肝炎ウイルス感染の治療のための方法及び組成物を提供する。場合により、ウイルス量を経時的にモニターしてウイルスリバウンドを調べてもよい。有意なウイルス量が再び観察されない限り治療を再開せず、観察されたときには1、2又は3日間の治療を繰り返す。この治療は患者の健康をモニターし、維持するために無期限に継続され得る。
【0046】
治療され得るフラビウイルスには、肝炎ウイルス属(HCV)、ペスチウイルス属(BVDV、CSFV、BDV)及びフラビウイルス属(デング熱ウイルス、西ナイルウイルスを含めた日本脳炎ウイルス群及び黄熱ウイルス)のすべてのメンバーが含まれ得る。
【0047】
別の実施態様において、より重篤なフラビウイルス感染の場合、ゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグを約50〜約1300mg/m2/日の範囲の用量で1日〜7日間投与した後治療を休止する。場合によりウイルス量を経時的にモニターしてウイルスのリバウンドを調べてもよい。有意なウイルス量が再び観察されない限り治療を再開せず、観察されたときには1〜7日間、より好ましくは1、2又は3日間の治療を繰り返す。この治療は患者の健康をモニターし、維持するために無期限に継続され得る。
【0048】
I.本発明の化合物
本発明の特定実施態様において、式:
【0049】
【化5】
を有するβ−D−ヌクレオシド、そのβ−L−エナンチオマー、その医薬的に許容され得る塩もしくはプロドラッグがフラビウイルス感染、特にHCV感染を治療または予防するために提供される。好ましい実施態様において、化合物はゲムシタビン、またはその医薬的に許容され得る塩、エステルもしくはプロドラッグである。前記化合物は、例えばその活性、バイオアベイラビリティ、安定性を修飾するため、またはヌクレオシドの諸特性を変化させるためにN4及び/または3’位及び/または5’位でアルキル化、アシル化または他の方法で誘導体化されていてもよい。こうすると、非静脈処方物をより安定性とし得る。1つの実施態様において、化合物はN4及び/または3’位及び/または5’位をバリンのようなアミノ酸でアシル化されている。
【0050】
本発明の包括的な側面において、活性化合物は一般式(I)を有するβ−Dもしくはβ−L−ヌクレオシド、またはその医薬的に許容され得る塩もしくはプロドラッグ(本明細書中、ゲムシタビン誘導体と称する)である。
【0051】
【化6】
【0052】
上記式中、
RはH、ハロゲン(F、Cl、Br、I)、OH、OR’、SH、SR’、NH2、NHR’、NR’2、C1−6低級アルキル、ハロゲン化(F、Cl、Br、I)C1−6低級アルキル(例えば、CF3及びCH2CH2F)、C2−6低級アルケニル(例えば、CH=CH2)、ハロゲン化(F、Cl、Br、I)C2−6低級アルケニル(例えば、CH=CHCl、CH=CHBr及びCH=CHI)、C2−6低級アルキニル(例えば、C≡CH)、ハロゲン化(F、Cl、Br、I)C2−6低級アルキニル、C1−6低級アルコキシ(例えば、CH2OH及びCH2CH2OH)、CO2H、CO2R’、CONH2、CONHR’、CONR’2、CH=CHCO2H、CH=CHCO2R’であり;
XはH、ハロゲン、OH、OR’、OCH3、SH、SR’、SCH3、NH2、NHR’、NR’2またはCH3であり;
各R’は独立して水素、C1−6低級アルキルまたはC1−6低級シクロアルキルであり;
ZはO、SまたはCH2であり;
R4はH、モノホスフェート、ジホスフェート、トリホスフェート、安定化ホスフェートプロドラッグ、アシル、アルキル、スルホネートエステル、脂質、リン脂質、アミノ酸、炭水化物、ペプチド、コレステロール、またはインビボ投与したときにR4がHまたはホスフェートである化合物を生じ得る他の医薬的に許容され得る離脱基であり、;
R3はFまたはOHである。
【0053】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはハロゲンである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0054】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはアルキルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0055】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはハロゲン化アルキルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0056】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはCH3であり、RはNH2である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0057】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはOR’であり、Rはハロゲンである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0058】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはハロゲンであり、R4は水素である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0059】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0060】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはアルケニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0061】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはアルキニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0062】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはハロゲン化アルケニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0063】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはハロゲン化アルキニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0064】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはアルコキシである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0065】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、RはCO2Hである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0066】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、RはCO2R’である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0067】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、RはCONH2である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0068】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、RはCONHR’である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0069】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、Rはハロゲンである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0070】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、Rはハロゲンである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0071】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、Rはアルキルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0072】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、Rはハロゲン化アルキルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0073】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはCH3であり、R3はOHであり、RはNH2である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0074】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはOR’であり、R3はOHであり、Rはハロゲンである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0075】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、Rはハロゲンであり、R4は水素である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0076】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0077】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはCH3であり、R3はFであり、Rはアルケニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0078】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはCH3であり、R3はOHであり、Rはアルキニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0079】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、Rはハロゲン化アルケニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0080】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、Rはハロゲン化アルキニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0081】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、Rはアルコキシである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0082】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、RはCO2Hである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0083】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、RはCO2R’である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0084】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、RはCONH2である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0085】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、RはCONHR’である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0086】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、Rはハロゲンである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0087】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNH2であり、R3はOHであり、Rはハロゲンであり、R4はHである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0088】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはSH2であり、R3はOHであり、Rはハロゲンである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0089】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNHR’であり、R3はOHであり、Rはハロゲンである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0090】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、Rはハロゲンである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0091】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、Rはアルキルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0092】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、Rはアルケニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0093】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、Rはアルキニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0094】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、Rはハロゲン化アルケニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0095】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、Rはハロゲン化アルキニルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0096】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、Rはアルコキシである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0097】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、RはCO2Hである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0098】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはOR’である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0099】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはNHR’である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0100】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、XはCONH2である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0101】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R3はFである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0102】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R3はOHである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0103】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R4はHである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0104】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R4はモノホスフェートである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0105】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R4はジホスフェートである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0106】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R4はトリホスフェートである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0107】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R4はアシルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0108】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R4はHであり、ZはOである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0109】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R4はHであり、ZはCH2である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0110】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、RはHであり、R3はFであり、R4はアシルである)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0111】
本発明の別の実施態様において、活性化合物は一般式(I)(式中、R4はHであり、RはOR’である)を有するβ−Dもしくはβ−L−ヌクレオシド、又はその医薬的に許容され得る塩もしくはプロドラッグであり、本明細書に更に記載されているその使用も提供する。
【0112】
定義
本明細書中、用語「アルキル」は、別段のことわりがない限り飽和の直鎖、分枝鎖または環状の1級、2級または3級炭化水素を指し、この中には非限定的にC1−16アルキルが含まれる。具体的には、メチル、エチル、プロピル、イソプロピル、シクロプロピル、ブチル、イソブチル、t−ブチル、ペンチル、シクロペンチル、イソペンチル、ネオペンチル、ヘキシル、イソヘキシル、シクロヘキシル、シクロヘキシルメチル、3−メチルペンチル、2,2−ジメチルブチル及び2,3−ジメチルブチルが含まれる。アルキル基は場合により、保護されていなくても所要により保護されていてもよい、アルキル、ハロ、ハロアルキル、ヒドロキシ、カルボキシル、アシル、アシルオキシ、アミノ、アミド(amido)、カルボキシル誘導体、アルキルアミノ、ジアルキルアミノ、アリールアミノ、アルコキシ、アリールオキシ、ニトロ、シアノ、アジド、チオール、イミン、スルホン酸、スルフェート、スルホニル、スルファニル、スルフィニル、スルファモイル、エステル、カルボン酸、アミド(amide)、ホスホニル、ホスフィニル、ホスホリル、ホスフィン、チオエステル、チオエーテル、酸ハライド、無水物、オキシム、ヒドロジン、カルバメート、ホスホン酸、ホスフェート、ホスホネート、及び本化合物の医薬活性を阻害しない任意の実行可能な官能基からなる群から選択される1つ以上の部分で置換されていてもよい。なお、保護基は援用により本明細書に含まれるとするGreeneら,「有機合成における保護基(Protective Groups in Organic Synthesis)」,第2版,John Wiley and Sons(1991年)発行に教示されているように当業者に公知である。
【0113】
本明細書中、用語「低級アルキル」は、別段のことわりがない限りC1−4飽和直鎖、分枝鎖、または適当ならば環状(例えば、シクロプロピル)アルキル基を指し、この基は未置換でも置換されていてもよい。
【0114】
本明細書中、用語「アルキレン」または「アルケニル」は、直鎖または分枝鎖構造を有する飽和ヒドロカルビルジイル基を指し、この中には非限定的に1〜10炭素原子を有するものが含まれる。この用語の範囲には、メチレン、1,2−エタン−ジイル、1,1−エタン−ジイル、1,3−プロパン−ジイル、1,2−プロパン−ジイル、1,3−ブタン−ジイル、1,4−ブタン−ジイル等が含まれる。アルキレン基または本明細書に記載されている他の2価部分は場合により、保護されていなくても所要により保護されていてもよい、アルキル、ハロ、ハロアルキル、ヒドロキシ、カルボキシル、アシル、アシルオキシ、アミノ、アミド、カルボキシル誘導体、アルキルアミノ、アジド、ジアルキルアミノ、アリールアミノ、アルコキシ、アリールオキシ、ニトロ、シアノ、スルホン酸、チオール、イミン、スルホニル、スルファニル、スルフィニル、スルファモイル、エステル、カルボン酸、アミド、ホスホニル、ホスフィニル、ホスホリル、ホスフィン、チオエステル、チオエーテル、酸ハライド、無水物、オキシム、ヒドロジン、カルバメート、ホスホン酸、ホスホネート、及び本化合物の医薬活性を阻害しない任意の実行可能な官能基からなる群から選択される1つ以上の部分で置換されていてもよい。なお、保護基は援用により本明細書に含まれるとするGreeneら,「有機合成における保護基(Protective Groups in Organic Synthesis)」,第2版,John Wiley and Sons(1991年)発行に教示されているように当業者に公知である。
【0115】
本明細書中、用語「アリール」は、別段のことわりがない限りフェニル、ビフェニルまたはナフチルを指し、好ましくはフェニルである。この用語は置換部分及び未置換部分を含む。アリール基は、保護されていなくても所要により保護されていてもよい、ブロモ、クロロ、フルオロ、ヨード、ヒドロキシ、アジド、アミノ、アルキルアミノ、アリールアミノ、アルコキシ、アリールオキシ、ニトロ、シアノ、スルホン酸、スルフェート、ホスホン酸、ホスフェート及びホスホネートからなる群から選択される1つ以上の部分で置換されていてもよい。なお、保護基は援用により本明細書に含まれるとするGreeneら,「有機合成における保護基(Protective Groups in Organic Synthesis)」,第2版,John Wiley and Sons(1991年)発行に教示されているように当業者に公知である。
【0116】
本明細書中、用語「アミノ酸」は、天然及び合成α、β、γまたはδ−アミノ酸を含み、その中には非限定的にアラニル、バリニル、ロイシニル、イソロイシニル、プロリニル、フェニルアラニニル、トリプトファニル、メチオニニル、グリシニル、セリニル、スレオニニル、システイニル、チロシニル、アスパラギニル、グルタミニル、アスパルトイル、グルタロイル、リシニル、アルギニニル、ヒスチジニル、β−アラニル、β−バリニル、β−ロイシニル、β−イソロイシニル、β−プロリニル、β−フェニルアラニニル、β−トリプトファニル、β−メチオニニル、β−グリシニル、β−セリニル、β−スレオニニル、β−システイニル、β−チロシニル、β−アスパラギニル、β−グルタミニル、β−アスパルトイル、β−グルタロイル、β−リシニル、β−アラギニニル及びβ−ヒスチジニルが含まれる。
【0117】
本明細書中、用語「アラルキル」は、別段のことわりがない限り上記アルキル基を介して分子に結合した上記アリール基を指す。本明細書中、用語「アルカリール」または「アルキルアリール」は、別段のことわりがない限り上記アリール基を介して分子に結合した上記アルキル基を指す。これらの各基においてアルキル基は場合により上記したように置換されていてもよく、アリール基は場合により、保護されていなくても所要により保護されていてもよい、アルキル、ハロ、ハロアルキル、ヒドロキシ、カルボキシル、アシル、アシルオキシ、アミノ、アミド、アジド、カルボキシル誘導体、アルキルアミノ、ジアルキルアミノ、アリールアミノ、アルコキシ、アリールオキシ、ニトロ、シアノ、スルホン酸、チオール、イミン、スルホニル、スルファニル、スルフィニル、スルファモイル、エステル、カルボン酸、アミド、ホスホニル、ホスフィニル、ホスホリル、ホスフィン、チオエステル、チオエーテル、酸ハライド、無水物、オキシム、ヒドロジン、カルバメート、ホスホン酸、ホスホネート、及び本化合物の医薬活性を阻害しない任意の実行可能な官能基からなる群から選択される1つ以上の部分で置換されていてもよい。なお、保護基は援用により本明細書に含まれるとするGreeneら,「有機合成における保護基(Protective Groups in Organic Synthesis)」,第2版,John Wiley and Sons(1991年)発行に教示されているように当業者に公知である。用語「アリール」の範囲内には、具体的にフェニル、ナフチル、フェニルメチル、フェニルエチル、3,4,5−トリヒドロキシフェニル、3,4,5−トリメトキシフェニル、3,4,5−トリエトキシフェニル、4−クロロフェニル、4−メチルフェニル、3,5−ジ−t−ブチル−4−ヒドロキシフェニル、4−フルオロフェニル、4−クロロ−1−ナフチル、2−メチル−1−ナフチルメチル、2−ナフチルメチル、4−クロロフェニルメチル、4−t−ブチルフェニル、4−t−ブチルフェニルメチル等が含まれる。
【0118】
用語「アルキルアミノ」または「アリールアミノ」は、それぞれ1または2個のアルキル置換基またはアリール置換基を有するアミノ基を指す。
【0119】
本明細書中、用語「ハロゲン」は、フッ素、塩素、臭素及びヨウ素を含む。
【0120】
用語「エニンチオマー的に富化された」は、本明細書中、ヌクレオシドであって、当該ヌクレオシドの単一エナンチオマーを少なくとも約95%、好ましくは少なくとも96%、より好ましくは少なくとも97%、更に好ましくは少なくとも98%、特に好ましくは少なくとも約99%以上含むものを指すべく使用される。好ましい実施態様において、ヌクレオシド類縁体はエナンチオマー的に富化された形態で提供される。
【0121】
本明細書中、用語「宿主」は、その中でウイルスが複製し得る単細胞または多細胞生物を指し、この中には細胞株及び動物が含まれ、好ましくはヒトである。或いは、宿主は複製または機能が本発明の化合物により変化を受け得るウイルスゲノムの一部を有していてもよい。用語「宿主」は具体的には、感染細胞、ウイルスゲノムの全部または一部をトランスフェクトした細胞及び動物、特に(チンパンジーを含めた)霊長類及びヒトを指す。異常細胞増殖に関して、用語「宿主」は異常細胞増殖を模倣し得る単細胞または多細胞生物を指す。用語「宿主」は具体的には、自然または不自然な原因により(例えば、それぞれ遺伝的変異または遺伝子工学操作により)異常に増殖する細胞及び動物、特に(チンパンジーを含めた)霊長類及びヒトを指す。本発明の多くの動物の用途において、宿主はヒト患者である。しかしながら、獣医学的用途(例えば、ウシにおけるウシウイルス性下痢症ウイルス、豚の豚コレラウイルス及びヒツジのボーダー病ウイルス)は本発明により明らかに予測される。
【0122】
用語「医薬的に許容され得る塩もしくはプロドラッグ」は、本明細書中、患者に投与したときに活性化合物を与える化合物の医薬的に許容され得るいずれもの形態(例えば、エステル、ホスフェートエステル、エステルまたは関連基の塩)を指すべく使用される。医薬的に許容され得る塩には、医薬的に許容され得る無機もしくは有機塩基または無機もしくは有機酸から誘導されるものが含まれる。適当な塩には、アルカリ金属(例えば、カリウム及びナトリウム)及びアルカリ土類金属(例えば、カルシウム及びマグネシウム)の他、製薬業界で公知の多数の他の酸から誘導されるものが含まれる。医薬的に許容され得るプロドラッグは宿主において代謝(例えば、加水分解または酸化)されて本発明の化合物を形成する化合物を指す。プロドラッグの典型例には、活性化合物の官能基部分上に生物学的に安定な保護基を有する化合物が含まれる。プロドラッグには、酸化、還元、アミノ化、脱アミノ化、ヒドロキシル化、脱ヒドロキシル化、加水分解、脱加水分解、アルキル化、脱アルキル化、アシル化、脱アシル化、リン酸化、脱リン酸化されると活性化合物を生じさせ得る化合物が含まれる。
【0123】
本発明の化合物はフラビウイルス科ウイルスに対して抗ウイルス活性を有するかもしくは異常細胞増殖に対して抗増殖活性を有し、または前記活性を呈する化合物に代謝される。
【0124】
立体異性及び多形
本発明の化合物は少なくとも2つのキラル中心を有し、光学的に活性な形態及びラセミ体形態で存在し得、単離され得る。幾つかの化合物は多型を示し得る。本発明は、本明細書に記載されている有用な特性を有する本発明の化合物のラセミ、光学活性、多型または立体異性体形態、またはそれらの混合物を包含する。光学活性形態は、例えばラセミ体形態を再結晶化技術、光学活性な出発物質からの合成、キラル合成、またはキラル固定相を用いるクロマトグラフィー分離、または酵素分割により製造され得る。
【0125】
化合物の光学的に活性な形態は、再結晶技術によるラセミ形態の分割、光学活性出発物質からの合成、キラル合成、またはキラル固定相を用いるクロマトグラフィー分離を含めた当業界で公知の方法を用いて製造され得る。
【0126】
光学的に活性な物質を得るための方法の例は以下の手順を少なくとも1つ含む。
i) 結晶の物理的分離:
各エナンチオマーの巨視的結晶を手動で分離する技術。この技術は、別のエナンチオマーの結晶が存在する場合、すなわち物質が集塊で、結晶が視覚的に認識できる場合に使用され得る。
ii) 同時結晶化:
個々のエナンチオマーをラセミ体溶液から別々に結晶化させる技術。ラセミ体が固体の集塊のときにのみ使用され得る。
iii) 酵素分割:
エナンチオマーに対する酵素との反応速度の違いを利用してラセミ体を部分的または完全に分離させる技術。
iv) 酵素的非対称合成:
所望エナンチオマーのエナンチオマー的に純粋なまたはエナンチオマー的に富化された合成前駆体を得るために合成の少なくとも1工程が酵素反応を利用している合成技術。
v) 化学非対称合成:
所望エナンチオマーを生成物中に非対称(すなわち、キラル性)を生ずる条件下でアキラル前駆体から合成する合成技術。非対称はキラル触媒またはキラル助剤を用いて達成され得る。
vi) ジアステレオマー分離:
ラセミ化合物を各エナンチオマーをジアステレオマーに変換するエナンチオマー的に純粋な試薬(キラル助剤)と反応させる技術。その後、生じたジアステレオマーをより区別し得る構造の違いを利用してクロマトグラフィーまたは結晶化により分離し、その後キラル助剤を除去して、所望エナンチオマーを得る。
vii) 一次及び二次非対称変換:
所望エナンチオマーからジアステレオマーが優先的に溶解するようにラセミ体からのジアステレオマーが平衡化するかまたは所望エナンチオマーからのジアステレオマーの優先的結晶化が最終的に原則としてすべての物質が所望エナンチオマーから結晶性ジアステレオマーに変換されるように平衡を混乱させる技術。その後、所望エナンチオマーをジアステレオマーから遊離させる。
viii) カイネティック分解:
この技術により、エナンチオマーのカイネティック条件下でのキラルな非ラセミ試薬または触媒との反応速度の違いを利用してラセミ体を部分的または完全に分割(または、部分的に分割した化合物の更なる分割)がなされる。
ix) 非ラセミ前駆体からのエナンチオ特異的合成:
所望エナンチオマーを非キラル出発物質から得、立体化学一体性が合成中全くまたは殆ど損なわれない技術。
x) キラル液体クロマトグラフィー:
ラセミ体のエナンチオマーを固定相との相互作用の違いを利用して(キラルHPLCを介するものを含む)液体移動相で分離する技術。固定相はキラル物質から構成され得、移動相は相互作用の違いを生じさせるために追加のキラル物質を含み得る。
xi) キラルガスクロマトグラフィー:
ラセミ体を気化させ、エナンチオマーを気体移動相での固定非ラセミキラル吸着剤相を含むカラムとの相互作用の違いを利用して分離する技術。
xii) キラル溶媒を用いる抽出:
1つのエナンチオマーを特定のキラル溶媒に優先的に溶解させることによりエナンチオマーを分離する技術。
xiii) キラル膜を介する移動:
ラセミ体を薄膜バリアと接触させる技術。前記バリアにより通常2つの混和性流体(一方はラセミ体を含む)が分離され、濃度または圧力差のような駆動力により膜バリアを優先的に移動する。分離は、ラセミ体の1つのエナンチオマーのみが通過し得る膜の非ラセミ体キラル性の結果起こる。
【0127】
模擬移動床クロマトグラフィーを含めたキラルクロマトグラフィーが1実施態様で使用される。各種のキラル固定相が市販されている。
【0128】
医薬組成物
式(I)を有する化合物、またはその医薬的に許容され得る塩もしくはプロドラッグに基づく医薬組成物は、フラビウイルス科ウイルスを処理するために治療有効量を場合により医薬的に許容され得る添加剤、担体または賦形剤と組み合わせて製造され得る。治療有効量は治療しようとする感染または状態、その重篤度、使用する治療方式、使用する化合物の薬物動態及び治療する患者に応じて異なり得る。
【0129】
本発明の1つの側面で、本発明の化合物を医薬的に許容され得る担体と混合して処方することが好ましい。通常、医薬組成物を静脈内形態で投与することが好ましいが、経口、非経口、筋肉内、経皮、頬腔、皮下、坐薬または他のルートを介して投与するようにも処方され得る。静脈内及び筋肉内処方物は好ましくは滅菌食塩液の形態で投与される。特定ルートに対する各種処方物を得るために当業者は本明細書の教示の範囲内で処方物を修飾することができる。特に、所望化合物を水または他のベヒクル中により可溶性とするための修飾は例えばルーチンの修飾(塩形成、エステル化等)により容易に実施し得る。
【0130】
経口処方物のような特定の剤形では、本発明の化合物のプロドラッグ形態、特にアシル化(アセチル化等)及びエーテル誘導体、ホスフェートエステルまたは塩形態を含めたプロドラッグ形態が好ましい。当業者は活性化合物の宿主生物または患者内の標的部位へのデリバリーを促進するために本発明の化合物をプロドラッグ形態に容易に修飾する方法を認識している。当業者は、(HCVを含めた)フラビウイルス科ウイルス感染の治療において化合物の所期効果を最大限とすべく所望化合物を宿主生物または患者の標的部位にデリバリーする際に適用可能ならばプロドラッグの有利な薬物動態パラメータも利用する。
【0131】
本発明の治療上活性な処方物中に含まれる化合物の量は(HCVを含めた)フラビウイルス科ウイルス感染を治療するために有効な量である。
【0132】
活性化合物は連続(静脈内滴注)投与しても数回(例えば、1日4回、1日2回等)経口投与してもよく、投与ルートの中には、他のルートの中で経口、局所、非経口、筋肉内、静脈内、皮下、経皮(浸透強化剤を含み得る)、頬腔及び坐薬投与が含まれ得る。経口投与ルートからの化合物のバイオアベイラビリティ及び安定性を高めるために腸溶性経口錠剤も使用し得る。最も有効な剤形は選択した特定化合物の薬物動態及び患者の病気の重篤度に依存する。
【0133】
第1実施態様において、本発明はフラビウイルス科ウイルス感染、特にC型肝炎ウイルス感染を治療するための方法及び組成物を提供し、その方法はゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグもしくは誘導体を約50〜約1300mg/m2/日の範囲の用量で1、2又は3日間投与した後治療を休止することを含む。別の実施態様では、より重篤なフラビウイルス科ウイルス感染に対してゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグもしくは誘導体を約50〜約1300mg/m2/日の用量で1日〜7日間(例えば、1、2、3、4、5、6または7日間)投与した後治療を休止する。
【0134】
本発明のゲムシタビンまたは別の活性化合物の1日用量は治療効果を最大限とするように選択され得る。非限定的用量の例は100〜1500mg/日、または200〜1000mg/日、特に300〜800mg/日である。
【0135】
化合物が安定な非毒性の酸性または塩基性塩を形成するのに十分な塩基性または酸性である場合には、前記化合物を医薬的に許容され得る塩として投与することも適当であり得る。医薬的に許容され得る塩には、医薬的に許容され得る無機もしくは有機塩基及び医薬的に許容され得る無機もしくは有機酸から誘導されるものが含まれる。適当な塩には、アルカリ金属(例えば、カリウム及びナトリウム)、アルカリ土類金属(例えば、カルシウム及びマグネシウム)の他に製薬業界で公知の酸が含まれる。特に、医薬的に許容され得る塩の例は、生理学的に許容され得るアニオンを形成する酸と形成される有機酸付加塩、例えばトシル酸塩、メタンスルホン酸塩、酢酸塩、クエン酸塩、マロン酸塩、酒石酸塩、コハク酸塩、安息香酸塩、アスコルビン酸塩、α−ケトグルタル酸塩及びα−グリセロリン酸塩である。適当な無機塩も形成され得、その中には硫酸塩、硝酸塩、炭酸水素酸塩、炭酸塩、塩酸塩及び臭化水素酸塩が含まれる。
【0136】
本明細書に記載されているいずれものヌクレオシドは、該ヌクレオシドの活性、バイオアベイラビリティや安定性を増加させるため、または諸特性を変化させるためにヌクレオチドプロドラッグとして投与され得る。多数のヌクレオシドプロドラッグリガンドが公知である。通常、ヌクレオシドのモノ−、ジ−またはトリホスフェートのアルキル化、アシル化または他の親油性修飾によりヌクレオチドの安定性が増加する。ホスフェート部分の1つ以上の水素原子を置換し得る置換基の例はアルキル、アリール、ステロイド、糖を含めた炭水化物、1,2−ジアシルグリセロール及びアルコールである。多くの置換基がR.Jones及びN.Bischofberger,Antiviral Research,27:1−17(1995)に記載されている。これらのいずれもが所望効果を達成するためにここに開示されているヌクレオシドと組み合わせて使用され得る。
【0137】
活性ヌクレオシドは、援用により本明細書に含まれるとする下表の参考文献に開示されているように5’−ホスホエーテル脂質または5’−エーテル脂質としても提供され得る。
【0138】
【表3】
好ましくはヌクレオシドの5’−OHでヌクレオシドに共役的に取り込まれ得る適当な親油性置換基または親油性調製物を開示している米国特許明細書の非限定例には、米国特許第5,149,794号明細書(Yatvinら,1992年9月22日)、同第5,194,654号明細書(Hostetlerら,1993年3月16日)、同第5,223,263号明細書(Hostetlerら,1993年6月29日)、同第5,256,641号明細書(Yatvinら,1993年10月26日)、同第5,411,947号明細書(Hostetlerら,1995年5月2日)、同第5,463,092号明細書(Hostetlerら,1995年10月31日)、同第5,543,389号明細書(Yatvinら,1996年8月6日)、同第5,543,390号明細書(Yatvinら,1996年8月6日)、同第5,543,391号明細書(Yatvinら,1996年8月6日)、同第5,554,728号明細書(Basavaら,1996年9月10日)が含まれ、これらはいずれも援用により本明細書に含まれるとする。本発明のヌクレオシドに結合し得る親油性置換基または親油性調製物を開示している外国特許文献には、国際特許出願公開第89/02733号パンフレット、同第90/00555号パンフレット、同第91/16920号パンフレット、同第91/18914号パンフレット、同第93/00910号パンフレット、同第94/26273号パンフレット、同第96/15132号パンフレット、欧州特許出願公開第0 350 287号明細書、欧州特許出願第93917054.4号明細書及び国際特許出願公開第91/19721号パンフレットが含まれる。
【0139】
本発明の医薬組成物を製造するためには、治療有効量の1つ以上の本発明の化合物を剤形を作成するための慣用の製薬調合技術に従って医薬的に許容され得る担体と混合することが好ましい。担体は投与、例えば静脈内または非経口投与するのに望ましい剤形に応じて各種形態をとり得る。経口剤形の医薬組成物を製造するには、通常の医薬用媒体を使用し得る。よって、懸濁液、エリキシル剤及び溶液のような液体経口組成物の場合には、水、グリコール、オイル、アルコール、矯臭剤、保存剤、着色剤等を含む適当な担体及び添加剤を使用し得る。散剤、錠剤、カプセル剤のような固体経口組成物、坐剤のような固体組成物の場合、スターチ、糖担体(例えば、デキストロース、マンニトール、ラクトース及び関連担体)、希釈剤、顆粒化剤、滑沢剤、結合剤、崩壊剤等を含む適当な担体及び添加剤を使用し得る。所望により、徐放性とするために錠剤またはカプセル剤に一般的技術により腸陽性コーティングを施してもよい。前記剤形を使用すると患者における化合物のバイオアベィラビリティが有意に影響され得る。
【0140】
非経口処方物の場合、担体は通常滅菌水または塩化ナトリウム水溶液を含む、分散を助けるものを含めた他の成分も配合され得る。滅菌水を使用し、滅菌物として維持しようとするときには、組成物も担体も滅菌しなければならない。注射用懸濁液も調製可能であるが、この場合適当な液体担体、懸濁剤等が使用され得る。
【0141】
(ウイルス抗原を標的とするリポソームを含めた)リポソーム懸濁液も医薬的に許容され得る担体を作成するために慣用の方法により調製され得る。これは本発明のヌクレオシド化合物の遊離ヌクレオシド、アシルヌクレオシドまたはホスフェートエステルプロドラッグ形態をデリバリーするために適当であり得る。
【0142】
加えて、本発明の化合物は1つ以上の抗ウイルス剤、抗−HIV剤、抗−HBV剤、抗−HCV剤、抗肝炎剤またはインターフェロン、抗癌剤、抗細菌剤、または他の化合物と一緒にまたは交互に投与され得る。好ましい化合物には、α−インターフェロン及びリバビリンが含まれる。本発明の特定化合物は他の化合物の代謝、異化または不活化を減らすことにより本発明の特定化合物の生物学的活性を高めるために有効であり得、よって所期目的のために同時投与され得る。
【0143】
追加実施態様では、場合により本明細書に開示されている医薬的に許容され得る担体または希釈剤中に含まれる医薬的有効量のゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグを、場合により1つ以上の他の抗ウイルス剤と一緒または交互に哺乳動物に投与することを含む、ウイルス関連疾患を患っている哺乳動物の治療または予防方法を提供する。好ましい実施態様において、哺乳動物はヒトである。
【0144】
特に、本発明は、肝炎ウイルス属(HCV)、ペスチウイルス属(BVDV、CSFV、BDV)またはフラビウイルス属(デング熱ウイルス、西ナイルウイルスを含めた日本脳炎ウイルス及び黄熱ウイルス)のすべてのメンバーを含めたフラビウイルス科ウイルス感染を治療または予防するための方法及びそのための使用を包含する。
【0145】
本発明を以下に更に説明する。本明細書中の実施例は本発明の理解を助けるために提示されている。実施例は請求の範囲に記載されている本発明を限定するものではなく、限定するものと解釈されるべきでない。
【0146】
フラビウイルス科ウイルス感染を治療するための治療法
抗ウイルス剤で長期間治療した後にウイルスの薬剤耐性変異株が出現し得ることは認められている。最も典型的には、薬剤耐性はウイルス複製サイクルで使用される酵素(最も典型的には、HCVの場合RNA依存性RNAポリメラーゼ)をコードする遺伝子の変異により生じる。ウイルス感染に対する薬物の効果は化合物を主薬物により生ずる変異とは異なる変異を誘発する第2及び多分第3抗ウイルス化合物と一緒または交互に投与することにより延長、増強または回復させることができることが立証されている。また、薬物の薬物動態、体内分布または他のパラメーターは前記した併用または交互治療により変化させることができる。通常、併用治療はウイルスに対して複数の同時ストレスを誘発するので、通常併用治療が交互治療よりも好ましい。
【0147】
フラビウイルス科ウイルス、特にC型肝炎ウイルスに対して活性であると同定され、よってゲムシタビン、その塩、プロドラッグまたは誘導体と一緒または交互に使用され得る薬物の例を下記する:
(1) インターフェロン及び/またはリバビリン。
【0148】
(2) α−ケトアミド及びヒドラジノウレアを含めた基質ベースのNS3プロテアーゼ阻害剤(Attwoodら,「抗ウイルスペプチド誘導体(Antiviral peptide derivatives)」,国際特許出願公開第98/22496号パンフレット(1998);Attwoodら,Antiviral Chemistry and Chemotherapy,10:259−273(1999);Attwoodら,「抗ウイルス剤としてのアミノ酸誘導体の製造及び使用(Preparation and use of amino acid derivatives as anti-viral agents)」,独国特許出願公開第19914474号明細書;Tungら,「セリンプロテアーゼ、特にC型肝炎ウイルスNS3プロテアーゼの阻害剤(Inhibitors of serine proteases, particularly hepatitis C virus NS3 protease)」,国際特許出願公開第98/17679号パンフレット、及びボロン酸またはホスホネートのような求電子を停止させる阻害剤(Llinas−Brunetら,「C型肝炎阻害剤ペプチドアナログ(Hepatitis C inhibitor peptide analogues)」,国際特許出願公開第99/07734号パンフレット)。
【0149】
(3) 2,4,6−トリヒドロキシ−3−ニトロ−ベンズアミド誘導体のような非基質ベースの阻害剤(K.Sudoら,Biochemical and Biophysical Research Communications,238:643−647(1997);K.Sudoら,Antiviral Chemistry and Chemotherapy,9:186(1998))。例えば、アミド上を14炭素鎖で置換したRD3−4082及びp−フェノキシフェニル基をプロセッシングするRD3−4078。
【0150】
(4) NS3/4A融合タンパク質及びNS5A/5B基質を用いる逆相HPLCアッセイで相対阻害を示すチアゾリジン誘導体(K.Sudoら,Antiviral Research,32:9−18(1996))、特に長アルキル鎖で置換された融合シンナモイル部分を有する化合物RD−1−6250、RD4 6205及びRD4 6193。
【0151】
(5) N.Kakiuchiら,J.EBS Letters,421:217−220;N.Takeshitaら,Analytical Biochemistry,247:242−246(1997)が同定したチアゾリジン及びベンズアニリド。
【0152】
(6) SDS−PAGE及びオートラジオグラフィーアッセイにおいてプロテアーゼに対して活性を有する、フェナン−スレンキノン。ストレプトミセス種の発酵培養ブロスから単離したSch 68631(M.Chuら,Tetrahedron Letters,37:7229−7232(1996))、真菌Penicillium griscofuluumから単離したSch 351633。これらはシンチレーションプロキシミティアッセイにおいて活性を示す(M.Chuら,Bioorganic and Medicinal Chemistry Letters,9:1949−1952))。
【0153】
(7) ヒルから単離した高分子elgin cをベースとする選択的N3阻害剤(M.A.Qasimら,Biochemistry,36:1598−1607(1997))。
【0154】
(8) ヘリカーゼ阻害剤(G.D.Dianaら,「C型肝炎治療のための化合物、組成物及び方法(Compounds, compositions and methods for treatment of hepatitis C)」,米国特許第5,633,358号明細書;G.D.Dianaら,「ピペリジン誘導体、その医薬組成物及びC型肝炎治療におけるその使用(Piperidine derivatives, pharmaceutical compositions thereof and their use in the treatment of hepatitis C)」,国際特許出願公開第97/36554号パンフレット)。
【0155】
(9) ポリメラーゼ阻害剤、例えばヌクレオチドアナログのグリオトキシン(R.Ferrariら,Jouranl of Virology,73:1649−1654(1999))及び天然産物のセルレニン(V.Lohmannら,Virology,249:108−118(1998))。
【0156】
(10) ウイルスの5’−非コード領域(NCR)中の配列ストレッチに相補的なアンチセンスホスホロチオエートオリゴデオキシヌクレオチド(S−ODN)(M.Altら,Hepatology,22:707−717(1995))、またはNCRの3’末端を含むヌクレオチド326−348及びHCV RNAのコアコード領域に位置するヌクレオチド371−388(M.Alaら,Archives of Virology,142:589−599(1997);U.Galderisi,Journal of Cellular Physiology,181:251−257(1999))。
【0157】
(11) IRES依存性翻訳の阻害剤(N.Ikedaら,「C型肝炎の予防及び治療剤(Agent for the prevention and treatment of hepatitis C)」,特開平08/268890号公報:Y.Kaiら,「ウイルス疾患の予防及び治療(Prevention and treatment of viral diseases)」,特開平10/101591号公報)。
【0158】
(12) ヌクレアーゼ耐性リボザイム(D.J.Maccjakら,Hepatology,30,abstract 995(1999))。
【0159】
(13) ヌクレオシド類縁体もフラビウイルス科ウイルス感染の治療のために開発された。
【0160】
(14) Idenix Pharmaceuticals,Ltd.の国際特許出願公開第01/90121号パンフレット(2001年5月23日出願)及び同第01/92282号パンフレット(2001年5月26日出願)は分枝鎖ヌクレオシド及びそのHCV、フラビウイルス及びペスチウイルスの治療における使用を開示している。Idenixパンフレットには、場合により医薬的に許容され得る担体中に含まれる有効量の生物学的に活性な1’,2’,3’または4’−分枝鎖β−Dまたはβ−L−ヌクレオシド、またはその医薬的に許容され得る塩もしくはプロドラッグを単独または組み合わせて投与することを含むヒト及び他の宿主動物におけるC型肝炎(及び、フラビウイルス及びペスチウイルス)感染の治療方法が開示されている。
【0161】
(15) Indenix Pharmaceuticals,Ltd.の国際特許出願公開第01/96353号パンフレット(2001年6月15日出願)はHBVの治療のための2’−デオキシ−β−L−ヌクレオシドの3’−プロドラッグを開示している。Beauchampの米国特許第4,957,924号明細書はアシクロビルの各種治療用エステルを開示している。
【0162】
(16) C型肝炎ウイルスを治療するために特定のヌクレオシド類縁体を使用することを開示している他の特許文献には、BioChem Pharma,Inc.(現Shire Biochem,Inc.)の国際特許出願公開第01/32153号パンフレット(PCT/CA00/01316,2000年11月3日出願)及び同第01/60315号パンフレット(PCT/CA01/00197,2001年2月19日出願);Merck & Co.,Inc.の国際特許出願公開第02/057425号パンフレット(PCT/US02/01531,2002年1月18日出願)及び同第02/057287号パンフレット(PCT/US02/03086,2002年1月18日出願);Rocheの国際特許出願公開第02/18404号パンフレット(PCT/EP01/09633,2001年8月21日出願);Pharmassetの国際特許出願公開第01/79246号パンフレット(2001年4月13日出願)及び同第02/32920号パンフレット(2001年10月18日出願)が含まれる。
【0163】
(17) 1−アミノ−アルキルシクロヘキサン(Goldらの米国特許第6,034,134号明細書)、アルキル脂質(Chojkierらの米国特許第5,922,757号明細書)、ビタミンE及び他の抗酸化剤(Chojkierらの米国特許第5,922,757号明細書)、スクアレン、アマンタジン、胆汁酸(Ozekiらの米国特許第5,846,964号明細書)、N−(ホスホノアセチル)−L−アスパラギン酸(Dianaらの米国特許第5,830,905号明細書)、ベンゼンジカルボキサミド(Dianaらの米国特許第5,633,388号明細書)、ポリアデニル酸誘導体(Wangらの米国特許第5,496,546号明細書)、2’,3’−ジデオキシイノシン(Yarchoanらの米国特許第5,026,687号明細書)及びベンズイミダゾール(Colacinoらの米国特許第5,891,874号明細書)を含めた他の各種化合物。
【0164】
(18) C型肝炎ウイルスの治療のために現在臨床開発されている他の化合物には、Schering−Ploughのインターロイキン−10、InterneuronのIP−50、VertexのMerimebodib VX−497、Endo Labs SolvayのAMANTADINE(Symmetrel)、RPIのHEPTAZYME、Idun Pharm.のIDN−6556、XTLのXTL−002、ChironのHCV/MF59、NABIのCIVACIR、ICNのLEVOVIRIN、ICNのVIRAMIDINE、Sci CloneのZADAXIN(チロシンα−1)、MaximのCEPLENE(ヒスタミン二塩酸塩)、Vertex/Eli LillyのVX950/LY570310、Isis Pharmaceutical/ElanのISIS 14803、Idun PharmaceuticalsのIDN−6556及びAKROS PharmaのJTK 003が含まれる。
【0165】
(19) エモリー大学及びジョージア大学研究財団の米国特許第6,348,587号明細書は、HIV、B型肝炎、C型肝炎及び異常細胞増殖の治療のための2’−フルオロヌクレオシドの使用を開示している。
【0166】
合成プロトコル
ピリミジンヌクレオシドの場合、ウリジン誘導体1(スキーム1)が出発物質であり、これを2,2’−無水誘導体2に変換し、無水ジオキサン中HFで処理する(Codingtonら,J.Org.Chem.,29:558(1964))。対応の2’−フルオロ−2’−デオキシウリジン誘導体3は40〜50%の収率で得られる。3の4位は各種方法により修飾され得る。2’−フルオロ−2’−デオキシシチジン誘導体4(R=R’=R”=H)は3からチオ化または塩素化を介する周知方法により容易に得ることができる。
【0167】
【化7】
【0168】
L−ウリジンから出発して、D−シリーズで合成したすべてのL−ヌクレオシド対応物を製造することができる。
【0169】
gem−ジフルオロヌクレオシドは、ナトリウム塩方法により2,2−ジフルオロ−1−O−アセチル−3,5−ジ−O−ベンゾイル−2−デオキシ−D−リボフラノース−2−ウロース8(スキーム2)を各種シリル化ピリミジン塩基またはプリンと縮合させることにより得ることができる。糖はReformatzky反応により2,3−O−イソプロピリデン−D−グリセラール5及びブロモジフルオロ酢酸エチル6から容易に製造され、その後保護基を酸で除去するとラクトン7が得られる。7をベンゾイル化した後DIBAL還元及びアセチル化することによりラクトンをラクトールに変換すると、8が生ずる。
【0170】
【化8】
【0171】
本発明の方法をより深く理解するために下記実施例を提示する。これらの実施例は例示にすぎず、本発明の範囲を限定することを意図するものではない。ここに記載の特定の溶媒、試薬または反応条件の代わりに同等、類似または適当な溶媒、試薬または反応条件を本発明の概念を逸脱することなく使用することができる。
【実施例1】
【0172】
フラビウイルスに対する候補化合物の抗ウイルス試験:
Huh7細胞におけるHCVレプリコン系。HCVレプリコンを有するHuh7細胞は、10% 胎仔ウシ血清、1X非必須アミノ酸、Pen−Strep−Glu(それぞれ100単位/L、100μg/L及び2.92mg/L)及び500〜1000μg/L G418を含有するDMEM培地(高グルコース、ピルビン酸塩なし)において培養し得る。抗ウイルススクリーニングアッセイは、G418を含まない同一培地において以下のように実施し得る。細胞をlog増殖相に維持するために細胞を96ウェルプレートに低密度で、例えば1000細胞/ウェルで接種する。細胞を接種した直後に試験化合物を添加し、インキュベータにおいて37℃で3〜7日間インキュベートする。その後、培地を除去し、細胞を総核酸抽出(レプリコンRNA及び宿主RNAを含む)のために作成する。次いで、レプリコンRNAをQ−RT−PCRプロトコルで増殖させ、定量し得る。レプリコンRNAの定量の観察された違いが試験化合物の抗ウイルス能力を表す1つの方法である。典型的な実験で、ネガティブコントロール及び無活性化合物環境では、かなりの量のレプリコンが産生される。このことは、両方の環境でHCV RT−PCRに対して測定された閾値サイクルが相互に近いので結論づけられ得る。前記実験で、化合物の抗ウイルス効果を表す1つの方法は試験化合物の閾値RT−PCRサイクルをネガティブコントロールの平均閾値RT−PCRサイクルで減ずることである。この値をデルタCt(ΔCtまたはDCt)と呼ぶ。3.3のΔCtはレプリコン産生の1−log低下に等しい(EC90に等しい)。2以上のΔCt値(レプリコンRNAの75%低下)でHCVレプリコンRNAレベルを低下させる化合物は抗ウイルス治療のための候補化合物である。前記候補化合物は一般式(I)を有する構造に属する。ポジティブコントロールとして、組換えインターフェロンα−2a(Roferon−A,米国ニュージャージー州に所在のHoffmann−Roche)をポジティブコントロールと一緒に用いる。
【0173】
しかしながら、このHCV ΔCt値はレプリコンコード化ウイルスRNA依存性RNAポリメラーゼの特異性パラメーターを含んでいない。典型的な環境で、化合物は宿主RNAポリメラーゼ活性及びレプリコンコード化ポリメラーゼ活性の両方を減少させ得る。従って、rRNA(または、他の宿主RNAポリメラーゼI産物)またはβ−アクチンmRNA(または、他の宿主RNAポリメラーゼII)の定量及び無薬物コントロールのRNAレベルとの比較が宿主RNAポリメラーゼに対する試験化合物の効果の相対評価である。
【0174】
HCV CΔtデータ及びrRNA ΔCtの両方を用いて、特異性パラメーターを導入し得る。このパラメーターは両方のΔCt値を相互に減ずることにより求められる。こうして、デルタ−デルタCt値(ΔΔCtまたはDDCt)が得られる。0よりも低い値はレプリコンコード化ポリメラーゼに対してより高い阻害効果があることを意味する。0以下のΔCt値は宿主rRNAレベルがレプリコンレベルよりも多く影響を受けることを意味する。概して、2以上のΔCt値は無薬物処置コントロールとは有意に異なり、認められる抗ウイルス活性を示すと見做される。しかしながら、2未満のΔCt値を有するが、限られた分子細胞毒性(rRNA ΔCT=0〜2)を有する化合物が可能性のある活性化合物である。
【0175】
別の典型的な環境で、化合物は宿主RNAポリメラーゼ活性を減少させるが、宿主DNAポリメラーゼ活性を減少させない。従って、rDNAまたはβ−アクチンDNA(または、他の宿主DNA断片)の定量及び無薬物コントロールのDNAレベルとの比較が試験化合物の細胞DNAポリメラーゼに対する阻害効果の相対評価である。
【0176】
HCV ΔCtデータ及びrDNA ΔCtを用いて、特異性パラメーターを導入し得る。このパラメーターは両方の白四角Ct値を相互に差し引くことにより求められる。こうして、ΔΔCt値が得られる。0以上の値はレプリコンコード化ポリメラーゼに対してより高い阻害効果があることを意味する。0よりも低いΔΔCt値は宿主rDNAレベルがレプリコンレベルよりも多く影響を受けることを意味する。概して、2以上のΔΔCt値は無薬物処置コントロールとは有意に異なると見做され、よって更なる評価に供される興味深い化合物である。しかしながら、2未満のΔΔCt値を有するが、限られた分子細胞毒性(rDNA ΔCT=0〜2)を有する化合物が望ましい。
【0177】
HCVレプリコンRNAレベルを特異的に減少させるが、細胞RNA及び/またはDNAレベルを僅かしか減少させない化合物が抗ウイルス治療のための候補化合物である。一般式(I)の群に属する候補化合物について(HCVを含めた)フラビウイルス科ウイルスRNAを減少させる特異的能力を評価し、有力な化合物を検出した。
【0178】
多くの研究がHCV遺伝子型1a及び1bは非タイプ1遺伝子型よりもα−インターフェロンをベースとする治療に対してより耐性であることを示している。このために、数人の医者はウイルス遺伝子型1aまたは1bに感染している患者に対して長い治療期間を処方していることがある。従って、1実施態様では、ゲムシタビンをHCV 1aまたは1bに感染している患者に対してウイルス量を減少させるのに有効な用量投与する。本発明の1実施態様では、ゲムシタビンをHCV 1aまたは1b遺伝子型を有する宿主に対してα−インターフェロンと別に投与する。更なる実施態様では、ゲムシタビンをHCV 1aまたは1b遺伝子型を有する宿主に対してα−インターフェロンと一緒に投与する。
【実施例2】
【0179】
ゲムシタビン(dFdC)の抗ウイルス活性
ゲムシタビンをDMSOに溶解し、これを自己複製HCV RNAを有するHuh7細胞の細胞モデル系の培地に0.1〜50dMの最終濃度で添加した。前記実験において化合物の抗ウイルス有効性を表す1つの方法は、試験化合物の閾値逆転写ポリメラーゼ連鎖反応(RT−PCR)サイクルをネガティブコントロールの平均閾値RT−PCRサイクルで減ずることである。この値はデルタCt(ΔCtまたはdCt)と呼ばれる。HCV ΔCtデータ及びrRNA ΔCtを利用して、特異性パラメーターが導入され得る。このパラメーターはΔCt値を相互から差し引くことにより求められる。こうして、デルタ−デルタCt値(ΔΔCtまたはddCt)が得られる。4日間インキュベートすると、レプリコンHCV RNAが用量依存的に減少した(図2)。3.3Ct値はレプリコンRNAの1−log低下に等しいので、EC90値は約70nMで達した。細胞DNAレベル(リボソームDNA)または細胞RNAレベル(リボソームRNA)の低下を更に分析したところ、宿主DNA及びRNAポリメラーゼに対するこの化合物の阻害能力を表すdCt(小)が生じた。これらの計算に基づいて、自己複製HCV RNAを有するHuh7細胞の細胞モデル系においてゲムシタビンはIC50(0.240μM)よりも低い濃度でHCV RNAレベルを有意に低下させた(EC50=0.040μM)。興味深いことに、不活性代謝物のdFdU(7.0μM)はHCVレリプリコン系においてdFdCに類似の活性を示した[dCT HCV=6.39、dCt rRNA=1.96及びddCt:4.42、(EC50及びIC50データは入手できず)]。
【実施例3】
【0180】
ヒトに1回治療後のゲムシタビンの抗ウイルス活性
HCVに感染した多病巣性HCC、肝硬変及び虚血性肝炎を呈している男性患者にオキサリプラチンと一緒にゲムシタビンHCl 1200mgを1000分間かけて投与した。耐性は許容でき、よって翌日患者に再びゲムシタビン約700mmgを投与した。第2回投与前のベースラインのウイルス量は6.49logコピー/mlであった。心臓に問題が生じたので、約700mgを投与後ゲムシタビンの注入を停止した。第2回投与から8時間後のHCV RNA測定値は4.04logコピー/mlであった。これは8時間で約2.5logの低下を示した。
【図面の簡単な説明】
【0181】
【図1】ゲムシタビン及びDNA修復の自己増強作用を示す。
【図2】ゲムシタビンを用いる治療に基づくレプリコンHCV RNAの用量依存的減少を示すグラフである(黒菱形:HCV RNAに対するΔCt)。このウイルス減少を細胞DNAレベル(リボソームDNA)または細胞RNAレベル(リボソームRNA)の減少と比較して、治療指数ΔΔCt値(黒三角:HCV−rDNA ΔΔCt、太字X:HCV−rRNA ΔΔCt)を求めた。
【特許請求の範囲】
【請求項1】
ゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグを
(i)50〜1300mg/m2−宿主表面積の量
(ii)連続1、2、3、4、5、6又は7日間毎日投与した後治療を休止する投薬計画で
投与することを含むC型肝炎ウイルス感染患者の治療方法。
【請求項2】
ゲムシタビン、またはその塩もしくはプロドラッグを200〜1000mg/m2/日の量投与する請求項1に記載の方法。
【請求項3】
ゲムシタビンを300〜800mg/m2/日の量投与する請求項1に記載の方法。
【請求項4】
投薬計画が1日1回1日間である請求項1に記載の方法。
【請求項5】
投薬計画が1日1回2日間である請求項1に記載の方法。
【請求項6】
投薬計画が1日1回3日間である請求項1に記載の方法。
【請求項7】
投薬計画が1日1回4日間である請求項1に記載の方法。
【請求項8】
投薬計画が1日1回5日間である請求項1に記載の方法。
【請求項9】
投薬計画が1日1回6日間である請求項1に記載の方法。
【請求項10】
投薬計画が1日1回7日間である請求項1に記載の方法。
【請求項11】
静脈投与する請求項1に記載の方法。
【請求項12】
治療を少なくとも2日間休止する請求項1に記載の方法。
【請求項13】
治療を少なくとも3日間休止する請求項1に記載の方法。
【請求項14】
治療を少なくとも1週間休止する請求項1に記載の方法。
【請求項15】
治療を少なくとも2週間休止する請求項1に記載の方法。
【請求項16】
治療を少なくとも3週間休止する請求項1に記載の方法。
【請求項17】
治療を少なくとも1ヶ月間休止する請求項1に記載の方法。
【請求項18】
ゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグを
(iii)50〜1300mg/m2−宿主表面積の量
(iv)連続1、2、3、4、5、6又は7日間毎日投与した後治療を休止する投薬計画で
投与することを含むフラビウイルス科ウイルス感染患者の治療方法。
【請求項19】
ゲムシタビン、またはその塩もしくはプロドラッグを200〜1000mg/m2/日の量投与する請求項18に記載の方法。
【請求項20】
ゲムシタビンを300〜800mg/m2/日の量投与する請求項18に記載の方法。
【請求項21】
投薬計画が1日1回1日間である請求項18に記載の方法。
【請求項22】
投薬計画が1日1回2日間である請求項18に記載の方法。
【請求項23】
投薬計画が1日1回3日間である請求項18に記載の方法。
【請求項24】
投薬計画が1日1回4日間である請求項18に記載の方法。
【請求項25】
投薬計画が1日1回5日間である請求項18に記載の方法。
【請求項26】
投薬計画が1日1回6日間である請求項18に記載の方法。
【請求項27】
投薬計画が1日1回7日間である請求項18に記載の方法。
【請求項28】
静脈投与する請求項18に記載の方法。
【請求項29】
治療を少なくとも2日間休止する請求項18に記載の方法。
【請求項30】
治療を少なくとも3日間休止する請求項18に記載の方法。
【請求項31】
治療を少なくとも1週間休止する請求項18に記載の方法。
【請求項32】
治療を少なくとも2週間休止する請求項18に記載の方法。
【請求項33】
治療を少なくとも3週間休止する請求項18に記載の方法。
【請求項34】
治療を少なくとも1ヶ月間休止する請求項18に記載の方法。
【請求項35】
50〜1300mg/m2の量の1種以上の他の抗ウイルス剤と共に、抗ウイルス有効量の構造式:
【化1】
[式中、
RはH、ハロゲン(F、Cl、Br、I)、OH、OR’、SH、SR’、NH2、NHR’、NR’2、C1−6低級アルキル、ハロゲン化(F、Cl、Br、I)C1−6低級アルキル(例えば、CF3及びCH2CH2F)、C2−6低級アルケニル(例えば、CH=CH2)、ハロゲン化(F、Cl、Br、I)C2−6低級アルケニル(例えば、CH=CHCl、CH=CHBr及びCH=CHI)、C2−6低級アルキニル(例えば、C≡CH)、ハロゲン化(F、Cl、Br、I)C2−6低級アルキニル、C1−6低級アルコキシ(例えば、CH2OH及びCH2CH2OH)、CO2H、CO2R’、CONH2、CONHR’、CONR’2、CH=CHCO2H、CH=CHCO2R’であり;
XはH、ハロゲン、OH、OR’、OCH3、SH、SR’、SCH3、NH2、NHR’、NR’2またはCH3であり;
各R’は独立して水素、C1−6低級アルキルまたはC1−6低級シクロアルキルであり;
ZはO、SまたはCH2であり;
R3はFまたはOHである]
を有するβ−Dもしくはβ−L−ヌクレオシド、またはその医薬的に許容され得る塩もしくはプロドラッグを
(i)50〜1300mg/m2−宿主表面積の量
(ii)連続1、2、3、4、5、6又は7日間毎日投与した後治療を休止する投薬計画で
投与することを含むフラビウイルス科ウイルスの治療方法。
【請求項36】
XがNH2であり、ZがOであり、R3がOHであり、RがHである請求項35に記載の方法。
【請求項37】
ゲムシタビン、またはその塩もしくはプロドラッグを200〜1000mg/m2/日の量投与する請求項35に記載の方法。
【請求項38】
ゲムシタビンを300〜800mg/m2/日の量投与する請求項35に記載の方法。
【請求項39】
投薬計画が1日1回1日間である請求項35に記載の方法。
【請求項40】
投薬計画が1日1回2日間である請求項35に記載の方法。
【請求項41】
投薬計画が1日1回3日間である請求項35に記載の方法。
【請求項42】
投薬計画が1日1回4日間である請求項35に記載の方法。
【請求項43】
投薬計画が1日1回5日間である請求項35に記載の方法。
【請求項44】
投薬計画が1日1回6日間である請求項35に記載の方法。
【請求項45】
投薬計画が1日1回7日間である請求項35に記載の方法。
【請求項46】
静脈投与する請求項35に記載の方法。
【請求項47】
治療を少なくとも2日間休止する請求項35に記載の方法。
【請求項48】
治療を少なくとも3日間休止する請求項35に記載の方法。
【請求項49】
治療を少なくとも1週間休止する請求項35に記載の方法。
【請求項50】
治療を少なくとも2週間休止する請求項35に記載の方法。
【請求項51】
治療を少なくとも3週間休止する請求項35に記載の方法。
【請求項52】
治療を少なくとも1ヶ月間休止する請求項35に記載の方法。
【請求項53】
フラビウイルス科ウイルスがC型肝炎ウイルスである請求項35に記載の方法。
【請求項54】
フラビウイルス科ウイルスが西ナイルウイルスである請求項18または35に記載の方法。
【請求項55】
フラビウイルス科ウイルスがデング熱ウイルスである請求項18または35に記載の方法。
【請求項56】
フラビウイルス科ウイルスがウシウイルス性下痢症ウイルスである請求項18または35に記載の方法。
【請求項57】
フラビウイルス科ウイルスがボーダー病ウイルスである請求項18または35に記載の方法。
【請求項58】
フラビウイルス科ウイルスが黄熱ウイルスである請求項18または35に記載の方法。
【請求項59】
C型肝炎ウイルス感染患者の治療における
(v)50〜1300mg/m2−宿主表面積の量
(vi)連続1、2、3、4、5、6又は7日間毎日投与した後治療を休止する投薬計画での
ゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグの使用。
【請求項60】
ゲムシタビン、またはその塩もしくはプロドラッグを200〜1000mg/m2/日の量投与する請求項59に記載の使用。
【請求項61】
ゲムシタビンを300〜800mg/m2/日の量投与する請求項59に記載の使用。
【請求項62】
投薬計画が1日1回1日間である請求項59に記載の使用。
【請求項63】
投薬計画が1日1回2日間である請求項59に記載の使用。
【請求項64】
投薬計画が1日1回3日間である請求項59に記載の使用。
【請求項65】
投薬計画が1日1回4日間である請求項59に記載の使用。
【請求項66】
投薬計画が1日1回5日間である請求項59に記載の使用。
【請求項67】
投薬計画が1日1回6日間である請求項59に記載の使用。
【請求項68】
投薬計画が1日1回7日間である請求項59に記載の使用。
【請求項69】
静脈投与する請求項59に記載の使用。
【請求項70】
治療を少なくとも2日間休止する請求項59に記載の使用。
【請求項71】
治療を少なくとも3日間休止する請求項59に記載の使用。
【請求項72】
治療を少なくとも1週間休止する請求項59に記載の使用。
【請求項73】
治療を少なくとも2週間休止する請求項59に記載の使用。
【請求項74】
治療を少なくとも3週間休止する請求項59に記載の使用。
【請求項75】
治療を少なくとも1ヶ月間休止する請求項59に記載の使用。
【請求項76】
フラビウイルス科ウイルス感染患者の治療のための
(vii)50〜1300mg/m2−宿主表面積の量
(viii)連続1、2、3、4、5、6又は7日間毎日投与した後治療を休止する投薬計画での
ゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグの使用。
【請求項77】
ゲムシタビン、またはその塩もしくはプロドラッグを200〜1000mg/m2/日の量投与する請求項76に記載の使用。
【請求項78】
ゲムシタビンを300〜800mg/m2/日の量投与する請求項76に記載の使用。
【請求項79】
投薬計画が1日1回1日間である請求項76に記載の使用。
【請求項80】
投薬計画が1日1回2日間である請求項76に記載の使用。
【請求項81】
投薬計画が1日1回3日間である請求項76に記載の使用。
【請求項82】
投薬計画が1日1回4日間である請求項76に記載の使用。
【請求項83】
投薬計画が1日1回5日間である請求項76に記載の使用。
【請求項84】
投薬計画が1日1回6日間である請求項76に記載の使用。
【請求項85】
投薬計画が1日1回7日間である請求項76に記載の使用。
【請求項86】
静脈投与する請求項76に記載の使用。
【請求項87】
治療を少なくとも2日間休止する請求項76に記載の使用。
【請求項88】
治療を少なくとも3日間休止する請求項76に記載の使用。
【請求項89】
治療を少なくとも1週間休止する請求項76に記載の使用。
【請求項90】
治療を少なくとも2週間休止する請求項76に記載の使用。
【請求項91】
治療を少なくとも3週間休止する請求項76に記載の使用。
【請求項92】
治療を少なくとも1ヶ月間休止する請求項76に記載の使用。
【請求項93】
フラビウイルスの治療のための、50〜1300mg/m2の量の1種以上の他の抗ウイルス剤と共に、抗ウイルス有効量の構造式:
【化2】
[式中、
RはH、ハロゲン(F、Cl、Br、I)、OH、OR’、SH、SR’、NH2、NHR’、NR’2、C1−6低級アルキル、ハロゲン化(F、Cl、Br、I)C1−6低級アルキル(例えば、CF3及びCH2CH2F)、C2−6低級アルケニル(例えば、CH=CH2)、ハロゲン化(F、Cl、Br、I)C2−6低級アルケニル(例えば、CH=CHCl、CH=CHBr及びCH=CHI)、C2−6低級アルキニル(例えば、C≡CH)、ハロゲン化(F、Cl、Br、I)C2−6低級アルキニル、C1−6低級アルコキシ(例えば、CH2OH及びCH2CH2OH)、CO2H、CO2R’、CONH2、CONHR’、CONR’2、CH=CHCO2H、CH=CHCO2R’であり;
XはH、ハロゲン、OH、OR’、OCH3、SH、SR’、SCH3、NH2、NHR’、NR’2またはCH3であり;
各R’は独立して水素、C1−6低級アルキルまたはC1−6低級シクロアルキルであり;
ZはO、SまたはCH2であり;
R3はFまたはOHである]
を有するβ−Dもしくはβ−L−ヌクレオシド、またはその医薬的に許容され得る塩もしくはプロドラッグの、
(i)50〜1300mg/m2−宿主表面積の量
(ii)連続1、2、3、4、5、6又は7日間毎日投与した後治療を休止する投薬計画での
使用。
【請求項94】
XがNH2であり、ZがOであり、R3がOHであり、RがHである請求項93に記載の使用。
【請求項95】
ゲムシタビン、またはその塩もしくはプロドラッグを200〜1000mg/m2/日の量投与する請求項93に記載の使用。
【請求項96】
ゲムシタビンを300〜800mg/m2/日の量投与する請求項93に記載の使用。
【請求項97】
投薬計画が1日1回1日間である請求項93に記載の使用。
【請求項98】
投薬計画が1日1回2日間である請求項93に記載の使用。
【請求項99】
投薬計画が1日1回3日間である請求項93に記載の使用。
【請求項100】
投薬計画が1日1回4日間である請求項93に記載の使用。
【請求項101】
投薬計画が1日1回5日間である請求項93に記載の使用。
【請求項102】
投薬計画が1日1回6日間である請求項93に記載の使用。
【請求項103】
投薬計画が1日1回7日間である請求項93に記載の使用。
【請求項104】
静脈投与する請求項93に記載の使用。
【請求項105】
治療を少なくとも2日間休止する請求項93に記載の使用。
【請求項106】
治療を少なくとも3日間休止する請求項93に記載の使用。
【請求項107】
治療を少なくとも1週間休止する請求項93に記載の使用。
【請求項108】
治療を少なくとも2週間休止する請求項93に記載の使用。
【請求項109】
治療を少なくとも3週間休止する請求項93に記載の使用。
【請求項110】
治療を少なくとも1ヶ月間休止する請求項93に記載の使用。
【請求項111】
フラビウイルス科ウイルスがC型肝炎ウイルスである請求項93に記載の使用。
【請求項112】
フラビウイルス科ウイルスが西ナイルウイルスである請求項76または93に記載の使用。
【請求項113】
フラビウイルス科ウイルスがデング熱ウイルスである請求項76または93に記載の使用。
【請求項114】
フラビウイルス科ウイルスがウシウイルス性下痢症ウイルスである請求項76または93に記載の使用。
【請求項115】
フラビウイルス科ウイルスがボーダー病ウイルスである請求項76または93に記載の使用。
【請求項116】
フラビウイルス科ウイルスが黄熱ウイルスである請求項76または93に記載の使用。
【請求項117】
フラビウイルス感染患者の治療薬剤の製造における
(ix)50〜1300mg/m2−宿主表面積の量
(x)連続1、2、3、4、5、6又は7日間毎日投与した後治療を休止する投薬計画での
ゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグの使用。
【請求項118】
フラビウイルス感染患者の治療薬剤の製造における
(xi)50〜1300mg/m2−宿主表面積の量
(xii)連続1、2、3、4、5、6又は7日間毎日投与した後治療を休止する投薬計画での
ゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグの使用。
【請求項119】
フラビウイルス科ウイルス治療薬剤の製造における、50〜1300mg/m2の量の1種以上の他の抗ウイルス剤と共に、抗ウイルス有効量の構造式:
【化3】
[式中、
RはH、ハロゲン(F、Cl、Br、I)、OH、OR’、SH、SR’、NH2、NHR’、NR’2、C1−6低級アルキル、ハロゲン化(F、Cl、Br、I)C1−6低級アルキル(例えば、CF3及びCH2CH2F)、C2−6低級アルケニル(例えば、CH=CH2)、ハロゲン化(F、Cl、Br、I)C2−6低級アルケニル(例えば、CH=CHCl、CH=CHBr及びCH=CHI)、C2−6低級アルキニル(例えば、C≡CH)、ハロゲン化(F、Cl、Br、I)C2−6低級アルキニル、C1−6低級アルコキシ(例えば、CH2OH及びCH2CH2OH)、CO2H、CO2R’、CONH2、CONHR’、CONR’2、CH=CHCO2H、CH=CHCO2R’であり;
XはH、ハロゲン、OH、OR’、OCH3、SH、SR’、SCH3、NH2、NHR’、NR’2またはCH3であり;
各R’は独立して水素、C1−6低級アルキルまたはC1−6低級シクロアルキルであり;
ZはO、SまたはCH2であり;
R3はFまたはOHである]
を有するβ−Dもしくはβ−L−ヌクレオシド、またはその医薬的に許容され得る塩もしくはプロドラッグの
(xi)50〜1300mg/m2−宿主表面積の量
(xii)連続1、2、3、4、5、6又は7日間毎日投与した後治療を休止する投薬計画での
使用。
【請求項1】
ゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグを
(i)50〜1300mg/m2−宿主表面積の量
(ii)連続1、2、3、4、5、6又は7日間毎日投与した後治療を休止する投薬計画で
投与することを含むC型肝炎ウイルス感染患者の治療方法。
【請求項2】
ゲムシタビン、またはその塩もしくはプロドラッグを200〜1000mg/m2/日の量投与する請求項1に記載の方法。
【請求項3】
ゲムシタビンを300〜800mg/m2/日の量投与する請求項1に記載の方法。
【請求項4】
投薬計画が1日1回1日間である請求項1に記載の方法。
【請求項5】
投薬計画が1日1回2日間である請求項1に記載の方法。
【請求項6】
投薬計画が1日1回3日間である請求項1に記載の方法。
【請求項7】
投薬計画が1日1回4日間である請求項1に記載の方法。
【請求項8】
投薬計画が1日1回5日間である請求項1に記載の方法。
【請求項9】
投薬計画が1日1回6日間である請求項1に記載の方法。
【請求項10】
投薬計画が1日1回7日間である請求項1に記載の方法。
【請求項11】
静脈投与する請求項1に記載の方法。
【請求項12】
治療を少なくとも2日間休止する請求項1に記載の方法。
【請求項13】
治療を少なくとも3日間休止する請求項1に記載の方法。
【請求項14】
治療を少なくとも1週間休止する請求項1に記載の方法。
【請求項15】
治療を少なくとも2週間休止する請求項1に記載の方法。
【請求項16】
治療を少なくとも3週間休止する請求項1に記載の方法。
【請求項17】
治療を少なくとも1ヶ月間休止する請求項1に記載の方法。
【請求項18】
ゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグを
(iii)50〜1300mg/m2−宿主表面積の量
(iv)連続1、2、3、4、5、6又は7日間毎日投与した後治療を休止する投薬計画で
投与することを含むフラビウイルス科ウイルス感染患者の治療方法。
【請求項19】
ゲムシタビン、またはその塩もしくはプロドラッグを200〜1000mg/m2/日の量投与する請求項18に記載の方法。
【請求項20】
ゲムシタビンを300〜800mg/m2/日の量投与する請求項18に記載の方法。
【請求項21】
投薬計画が1日1回1日間である請求項18に記載の方法。
【請求項22】
投薬計画が1日1回2日間である請求項18に記載の方法。
【請求項23】
投薬計画が1日1回3日間である請求項18に記載の方法。
【請求項24】
投薬計画が1日1回4日間である請求項18に記載の方法。
【請求項25】
投薬計画が1日1回5日間である請求項18に記載の方法。
【請求項26】
投薬計画が1日1回6日間である請求項18に記載の方法。
【請求項27】
投薬計画が1日1回7日間である請求項18に記載の方法。
【請求項28】
静脈投与する請求項18に記載の方法。
【請求項29】
治療を少なくとも2日間休止する請求項18に記載の方法。
【請求項30】
治療を少なくとも3日間休止する請求項18に記載の方法。
【請求項31】
治療を少なくとも1週間休止する請求項18に記載の方法。
【請求項32】
治療を少なくとも2週間休止する請求項18に記載の方法。
【請求項33】
治療を少なくとも3週間休止する請求項18に記載の方法。
【請求項34】
治療を少なくとも1ヶ月間休止する請求項18に記載の方法。
【請求項35】
50〜1300mg/m2の量の1種以上の他の抗ウイルス剤と共に、抗ウイルス有効量の構造式:
【化1】
[式中、
RはH、ハロゲン(F、Cl、Br、I)、OH、OR’、SH、SR’、NH2、NHR’、NR’2、C1−6低級アルキル、ハロゲン化(F、Cl、Br、I)C1−6低級アルキル(例えば、CF3及びCH2CH2F)、C2−6低級アルケニル(例えば、CH=CH2)、ハロゲン化(F、Cl、Br、I)C2−6低級アルケニル(例えば、CH=CHCl、CH=CHBr及びCH=CHI)、C2−6低級アルキニル(例えば、C≡CH)、ハロゲン化(F、Cl、Br、I)C2−6低級アルキニル、C1−6低級アルコキシ(例えば、CH2OH及びCH2CH2OH)、CO2H、CO2R’、CONH2、CONHR’、CONR’2、CH=CHCO2H、CH=CHCO2R’であり;
XはH、ハロゲン、OH、OR’、OCH3、SH、SR’、SCH3、NH2、NHR’、NR’2またはCH3であり;
各R’は独立して水素、C1−6低級アルキルまたはC1−6低級シクロアルキルであり;
ZはO、SまたはCH2であり;
R3はFまたはOHである]
を有するβ−Dもしくはβ−L−ヌクレオシド、またはその医薬的に許容され得る塩もしくはプロドラッグを
(i)50〜1300mg/m2−宿主表面積の量
(ii)連続1、2、3、4、5、6又は7日間毎日投与した後治療を休止する投薬計画で
投与することを含むフラビウイルス科ウイルスの治療方法。
【請求項36】
XがNH2であり、ZがOであり、R3がOHであり、RがHである請求項35に記載の方法。
【請求項37】
ゲムシタビン、またはその塩もしくはプロドラッグを200〜1000mg/m2/日の量投与する請求項35に記載の方法。
【請求項38】
ゲムシタビンを300〜800mg/m2/日の量投与する請求項35に記載の方法。
【請求項39】
投薬計画が1日1回1日間である請求項35に記載の方法。
【請求項40】
投薬計画が1日1回2日間である請求項35に記載の方法。
【請求項41】
投薬計画が1日1回3日間である請求項35に記載の方法。
【請求項42】
投薬計画が1日1回4日間である請求項35に記載の方法。
【請求項43】
投薬計画が1日1回5日間である請求項35に記載の方法。
【請求項44】
投薬計画が1日1回6日間である請求項35に記載の方法。
【請求項45】
投薬計画が1日1回7日間である請求項35に記載の方法。
【請求項46】
静脈投与する請求項35に記載の方法。
【請求項47】
治療を少なくとも2日間休止する請求項35に記載の方法。
【請求項48】
治療を少なくとも3日間休止する請求項35に記載の方法。
【請求項49】
治療を少なくとも1週間休止する請求項35に記載の方法。
【請求項50】
治療を少なくとも2週間休止する請求項35に記載の方法。
【請求項51】
治療を少なくとも3週間休止する請求項35に記載の方法。
【請求項52】
治療を少なくとも1ヶ月間休止する請求項35に記載の方法。
【請求項53】
フラビウイルス科ウイルスがC型肝炎ウイルスである請求項35に記載の方法。
【請求項54】
フラビウイルス科ウイルスが西ナイルウイルスである請求項18または35に記載の方法。
【請求項55】
フラビウイルス科ウイルスがデング熱ウイルスである請求項18または35に記載の方法。
【請求項56】
フラビウイルス科ウイルスがウシウイルス性下痢症ウイルスである請求項18または35に記載の方法。
【請求項57】
フラビウイルス科ウイルスがボーダー病ウイルスである請求項18または35に記載の方法。
【請求項58】
フラビウイルス科ウイルスが黄熱ウイルスである請求項18または35に記載の方法。
【請求項59】
C型肝炎ウイルス感染患者の治療における
(v)50〜1300mg/m2−宿主表面積の量
(vi)連続1、2、3、4、5、6又は7日間毎日投与した後治療を休止する投薬計画での
ゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグの使用。
【請求項60】
ゲムシタビン、またはその塩もしくはプロドラッグを200〜1000mg/m2/日の量投与する請求項59に記載の使用。
【請求項61】
ゲムシタビンを300〜800mg/m2/日の量投与する請求項59に記載の使用。
【請求項62】
投薬計画が1日1回1日間である請求項59に記載の使用。
【請求項63】
投薬計画が1日1回2日間である請求項59に記載の使用。
【請求項64】
投薬計画が1日1回3日間である請求項59に記載の使用。
【請求項65】
投薬計画が1日1回4日間である請求項59に記載の使用。
【請求項66】
投薬計画が1日1回5日間である請求項59に記載の使用。
【請求項67】
投薬計画が1日1回6日間である請求項59に記載の使用。
【請求項68】
投薬計画が1日1回7日間である請求項59に記載の使用。
【請求項69】
静脈投与する請求項59に記載の使用。
【請求項70】
治療を少なくとも2日間休止する請求項59に記載の使用。
【請求項71】
治療を少なくとも3日間休止する請求項59に記載の使用。
【請求項72】
治療を少なくとも1週間休止する請求項59に記載の使用。
【請求項73】
治療を少なくとも2週間休止する請求項59に記載の使用。
【請求項74】
治療を少なくとも3週間休止する請求項59に記載の使用。
【請求項75】
治療を少なくとも1ヶ月間休止する請求項59に記載の使用。
【請求項76】
フラビウイルス科ウイルス感染患者の治療のための
(vii)50〜1300mg/m2−宿主表面積の量
(viii)連続1、2、3、4、5、6又は7日間毎日投与した後治療を休止する投薬計画での
ゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグの使用。
【請求項77】
ゲムシタビン、またはその塩もしくはプロドラッグを200〜1000mg/m2/日の量投与する請求項76に記載の使用。
【請求項78】
ゲムシタビンを300〜800mg/m2/日の量投与する請求項76に記載の使用。
【請求項79】
投薬計画が1日1回1日間である請求項76に記載の使用。
【請求項80】
投薬計画が1日1回2日間である請求項76に記載の使用。
【請求項81】
投薬計画が1日1回3日間である請求項76に記載の使用。
【請求項82】
投薬計画が1日1回4日間である請求項76に記載の使用。
【請求項83】
投薬計画が1日1回5日間である請求項76に記載の使用。
【請求項84】
投薬計画が1日1回6日間である請求項76に記載の使用。
【請求項85】
投薬計画が1日1回7日間である請求項76に記載の使用。
【請求項86】
静脈投与する請求項76に記載の使用。
【請求項87】
治療を少なくとも2日間休止する請求項76に記載の使用。
【請求項88】
治療を少なくとも3日間休止する請求項76に記載の使用。
【請求項89】
治療を少なくとも1週間休止する請求項76に記載の使用。
【請求項90】
治療を少なくとも2週間休止する請求項76に記載の使用。
【請求項91】
治療を少なくとも3週間休止する請求項76に記載の使用。
【請求項92】
治療を少なくとも1ヶ月間休止する請求項76に記載の使用。
【請求項93】
フラビウイルスの治療のための、50〜1300mg/m2の量の1種以上の他の抗ウイルス剤と共に、抗ウイルス有効量の構造式:
【化2】
[式中、
RはH、ハロゲン(F、Cl、Br、I)、OH、OR’、SH、SR’、NH2、NHR’、NR’2、C1−6低級アルキル、ハロゲン化(F、Cl、Br、I)C1−6低級アルキル(例えば、CF3及びCH2CH2F)、C2−6低級アルケニル(例えば、CH=CH2)、ハロゲン化(F、Cl、Br、I)C2−6低級アルケニル(例えば、CH=CHCl、CH=CHBr及びCH=CHI)、C2−6低級アルキニル(例えば、C≡CH)、ハロゲン化(F、Cl、Br、I)C2−6低級アルキニル、C1−6低級アルコキシ(例えば、CH2OH及びCH2CH2OH)、CO2H、CO2R’、CONH2、CONHR’、CONR’2、CH=CHCO2H、CH=CHCO2R’であり;
XはH、ハロゲン、OH、OR’、OCH3、SH、SR’、SCH3、NH2、NHR’、NR’2またはCH3であり;
各R’は独立して水素、C1−6低級アルキルまたはC1−6低級シクロアルキルであり;
ZはO、SまたはCH2であり;
R3はFまたはOHである]
を有するβ−Dもしくはβ−L−ヌクレオシド、またはその医薬的に許容され得る塩もしくはプロドラッグの、
(i)50〜1300mg/m2−宿主表面積の量
(ii)連続1、2、3、4、5、6又は7日間毎日投与した後治療を休止する投薬計画での
使用。
【請求項94】
XがNH2であり、ZがOであり、R3がOHであり、RがHである請求項93に記載の使用。
【請求項95】
ゲムシタビン、またはその塩もしくはプロドラッグを200〜1000mg/m2/日の量投与する請求項93に記載の使用。
【請求項96】
ゲムシタビンを300〜800mg/m2/日の量投与する請求項93に記載の使用。
【請求項97】
投薬計画が1日1回1日間である請求項93に記載の使用。
【請求項98】
投薬計画が1日1回2日間である請求項93に記載の使用。
【請求項99】
投薬計画が1日1回3日間である請求項93に記載の使用。
【請求項100】
投薬計画が1日1回4日間である請求項93に記載の使用。
【請求項101】
投薬計画が1日1回5日間である請求項93に記載の使用。
【請求項102】
投薬計画が1日1回6日間である請求項93に記載の使用。
【請求項103】
投薬計画が1日1回7日間である請求項93に記載の使用。
【請求項104】
静脈投与する請求項93に記載の使用。
【請求項105】
治療を少なくとも2日間休止する請求項93に記載の使用。
【請求項106】
治療を少なくとも3日間休止する請求項93に記載の使用。
【請求項107】
治療を少なくとも1週間休止する請求項93に記載の使用。
【請求項108】
治療を少なくとも2週間休止する請求項93に記載の使用。
【請求項109】
治療を少なくとも3週間休止する請求項93に記載の使用。
【請求項110】
治療を少なくとも1ヶ月間休止する請求項93に記載の使用。
【請求項111】
フラビウイルス科ウイルスがC型肝炎ウイルスである請求項93に記載の使用。
【請求項112】
フラビウイルス科ウイルスが西ナイルウイルスである請求項76または93に記載の使用。
【請求項113】
フラビウイルス科ウイルスがデング熱ウイルスである請求項76または93に記載の使用。
【請求項114】
フラビウイルス科ウイルスがウシウイルス性下痢症ウイルスである請求項76または93に記載の使用。
【請求項115】
フラビウイルス科ウイルスがボーダー病ウイルスである請求項76または93に記載の使用。
【請求項116】
フラビウイルス科ウイルスが黄熱ウイルスである請求項76または93に記載の使用。
【請求項117】
フラビウイルス感染患者の治療薬剤の製造における
(ix)50〜1300mg/m2−宿主表面積の量
(x)連続1、2、3、4、5、6又は7日間毎日投与した後治療を休止する投薬計画での
ゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグの使用。
【請求項118】
フラビウイルス感染患者の治療薬剤の製造における
(xi)50〜1300mg/m2−宿主表面積の量
(xii)連続1、2、3、4、5、6又は7日間毎日投与した後治療を休止する投薬計画での
ゲムシタビン、またはその医薬的に許容され得る塩もしくはプロドラッグの使用。
【請求項119】
フラビウイルス科ウイルス治療薬剤の製造における、50〜1300mg/m2の量の1種以上の他の抗ウイルス剤と共に、抗ウイルス有効量の構造式:
【化3】
[式中、
RはH、ハロゲン(F、Cl、Br、I)、OH、OR’、SH、SR’、NH2、NHR’、NR’2、C1−6低級アルキル、ハロゲン化(F、Cl、Br、I)C1−6低級アルキル(例えば、CF3及びCH2CH2F)、C2−6低級アルケニル(例えば、CH=CH2)、ハロゲン化(F、Cl、Br、I)C2−6低級アルケニル(例えば、CH=CHCl、CH=CHBr及びCH=CHI)、C2−6低級アルキニル(例えば、C≡CH)、ハロゲン化(F、Cl、Br、I)C2−6低級アルキニル、C1−6低級アルコキシ(例えば、CH2OH及びCH2CH2OH)、CO2H、CO2R’、CONH2、CONHR’、CONR’2、CH=CHCO2H、CH=CHCO2R’であり;
XはH、ハロゲン、OH、OR’、OCH3、SH、SR’、SCH3、NH2、NHR’、NR’2またはCH3であり;
各R’は独立して水素、C1−6低級アルキルまたはC1−6低級シクロアルキルであり;
ZはO、SまたはCH2であり;
R3はFまたはOHである]
を有するβ−Dもしくはβ−L−ヌクレオシド、またはその医薬的に許容され得る塩もしくはプロドラッグの
(xi)50〜1300mg/m2−宿主表面積の量
(xii)連続1、2、3、4、5、6又は7日間毎日投与した後治療を休止する投薬計画での
使用。
【図1】

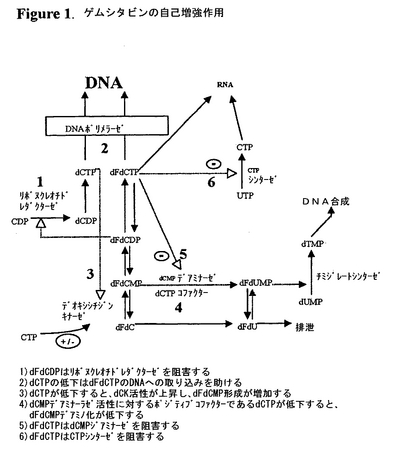
【図2】
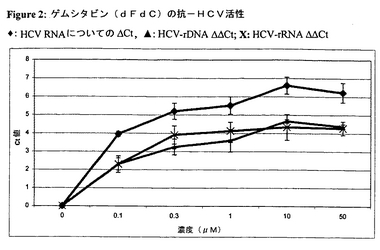
【図2】
【公表番号】特表2006−505490(P2006−505490A)
【公表日】平成18年2月16日(2006.2.16)
【国際特許分類】
【出願番号】特願2003−567349(P2003−567349)
【出願日】平成15年2月14日(2003.2.14)
【国際出願番号】PCT/US2003/004481
【国際公開番号】WO2003/068164
【国際公開日】平成15年8月21日(2003.8.21)
【出願人】(504310489)ファーマセット エルティーディー. (1)
【Fターム(参考)】
【公表日】平成18年2月16日(2006.2.16)
【国際特許分類】
【出願日】平成15年2月14日(2003.2.14)
【国際出願番号】PCT/US2003/004481
【国際公開番号】WO2003/068164
【国際公開日】平成15年8月21日(2003.8.21)
【出願人】(504310489)ファーマセット エルティーディー. (1)
【Fターム(参考)】
[ Back to top ]