
スピカマイシン誘導体を有するリポソーム製剤
【課題】水難溶性スピカマイシン誘導体(KRN5500)を安定に保持することができ、膜構成成分であるリン脂質と他の脂質の組成比や、リン脂質のアシル鎖長をコントロールすることで、血中滞留性に優れながらも、患部では効率よくKRN5500を放出することが可能なリポソーム製剤を提供する。
【解決手段】コレステロールが0〜50mol%および飽和脂肪酸のアシル鎖の鎖長がC14〜C18のいずれかであるリン脂質100〜50mol%を含有するリポソームに、化学式(I)で示されるスピカマイシン誘導体を有するリポソーム製剤:
【化1】

(式中、R1=H、R2=OHであって、Rは炭素数9〜15の直鎖のもしくは分岐したアルキルまたは炭素数10〜17の直鎖のアルケニルである。)
【解決手段】コレステロールが0〜50mol%および飽和脂肪酸のアシル鎖の鎖長がC14〜C18のいずれかであるリン脂質100〜50mol%を含有するリポソームに、化学式(I)で示されるスピカマイシン誘導体を有するリポソーム製剤:
【化1】
(式中、R1=H、R2=OHであって、Rは炭素数9〜15の直鎖のもしくは分岐したアルキルまたは炭素数10〜17の直鎖のアルケニルである。)
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、薬剤として水難溶性スピカマイシン誘導体を含有するリポソーム製剤に関するものである。
【背景技術】
【0002】
癌治療に用いられる抗癌剤は、癌細胞に対する高分子合成阻害活性のパターンから、DNA、RNA、蛋白合成阻害剤に分類される。主に、DNA、RNA合成に作用する抗癌剤の開発が主流であり、この場合は骨髄抑制など限られた器官への毒性が用量制限毒性(DLT)となる例が多い。一方、蛋白合成阻害剤は、肝臓をはじめとする生体維持に必要な重要な蛋白を合成する臓器に毒性が認められることが多く、こういった毒性が、薬効の効果が臨床で認められる前に発現するため、臨床で開発中止になる例が多い。このため、蛋白合成阻害剤は、より高い腫瘍細胞と正常細胞との選択毒性が要求される。
【0003】
式(I)で表されるスピカマイシン[式中、R1=H、R2=OHであって、Rは炭素数9〜15の直鎖のもしくは分岐したアルキルまたは炭素数10〜17の直鎖のアルケニルである]は、
【化1】
HL60細胞から分化誘導された抗腫瘍物質であり、蛋白合成阻害剤として見出された(特許文献1参照)。プリンの6位のアミノ基に特異なアミノヘプトース(以後、このアミノヘプトースをスピカミンと略称する)が結合し、このスピカミンの4位のアミノ基にグリシンがアミド結合し、更にこのグリシンのアミノ基に脂肪酸がアミド結合した構造を有している。該スピカマイシンを抗腫瘍剤として臨床的に使用できうる様に、より毒性が低く、より治療係数が高く、成分的に単一化されたスピカマイシン化合物の提供を目的に、脂肪酸側鎖部分の異なる種々のスピカマイシン誘導体の開発が行われてきた。その結果、これらの各種誘導体において脂肪酸部分の構造が最も抗腫瘍活性に寄与することが見出されており、スピカマイシンX(WO 90/15811号公開公報)(特許願PCT/JP90/00781号)ならびにKRN5500(6−[4−deoxy−4−(2E,4E)−tetradecadienoylglycyl]−amino−L−glycero−b−L−mannnoheptopyranosyl)amino−9H−purine)が、その目的に適合しうる抗腫瘍物質であることが報告されている(特許文献2参照)。
【0004】
これらの中でも特に式(II)で表されるKRN5500は、
【化2】
最も大きな治療指数を有するスピカマイシン誘導体であり、その効果は癌細胞の蛋白合成を阻害することで発揮されるため、新しい抗腫瘍メカニズムを有する新規の抗癌剤として開発が試みられた(非特許文献1)。上記KRN5500は、脂肪酸側鎖部分と細胞とのインターラクションが強力であるため、容易にインターナライゼーションが起こり、細胞内に取り込まれる。細胞内に取り込まれると、この脂肪酸側鎖は細胞膜などにある酵素によって、脂肪酸部分のみが加水分解されて活性本体のSAN−Gly(4−N−glycylspicamycin aminonucleoside)が産生される(非特許文献2)。各細胞がもっているKRN5500のSAN−Glyへの変換活性と、KRN5500のその細胞への殺細胞効果は相関することが認められており、正常細胞ではSAN−Glyへの変換活性は低く、KRN5500が強い殺細胞効果を示す癌細胞株では高く、このことから、KRN5500は比較的癌細胞選択性を有していることが報告されている(非特許文献3)。さらに、KRN5500はアドリアマイシン、ビンクリスチン、マイトマイシンC耐性細胞に対しても親株と同程度の殺細胞効果を示し、ほとんどのP-糖蛋白による薬剤排出機構を免れて多剤耐性細胞に対して有効であることも見出されている。
【0005】
このように、KRN5500は蛋白合成阻害という新しい抗腫瘍メカニズムを有し、かつ既存の抗癌剤の耐性を獲得した癌に対しても高い有効性が期待されるため、新規抗癌剤としての期待度が高く、国内、米国ともに臨床試験が行われた(非特許文献4、非特許文献5、非特許文献6)。しかしながら、国内、米国での第I相臨床試験ではそれぞれに重度の副作用として肺障害、肝障害が出現し、これらの副作用が薬剤の効果が臨床で認められる前に発現したため、開発の中断を余儀なくされている。
KRN5500の副作用に付随する課題の一つはその水難溶性にあり、水への溶解度が著しく低いため有機溶媒またはヒマシ油(クレモホールEL)の使用が余儀なくされている。そのため、KRN5500の薬剤自体の毒性に加えて溶媒毒性による副作用が発現する危険性があり、臨床試験で観察された副作用もこれらの毒性が関与していることが報告されている。また、有機溶媒の使用により製剤のpHが10−11と高いアルカリ性となっているため、臨床試験では中心静脈からの投与を余儀なくされており、そのため投与時間も長く、非常に扱いづらいことも報告されている。このようにKRN5500は薬剤自体の毒性の他に、溶解剤による毒性の問題があり、未だ解決すべき多くの課題があるため、臨床上使用されていないのが現状である。
【0006】
一般的に水難溶性薬物は、このような溶剤毒性による副作用を回避するためにDDS化の検討が試みられている。 例えば、水難溶性薬物としてパクリタキセルが挙げられるが、現在、注射液として投与するために溶媒にクレモホールEL(ポリオキシエチレンヒマシ油)とエタノールを用いている。このクレモホールELは過敏反応を引き起こすことが知られており、アナフィラキシー反応を引き起こし、呼吸困難、顔面紅潮、全身性の発疹、胸痛、頻脈、血圧低下、血管浮腫、蕁麻疹などの発現を認めることがある。そのため、この重篤な過敏症を防ぐためにパクリタキセルの投与時にステロイド剤や坑ヒスタミン薬の前投与薬が必要とされ、大変使いづらいのが現状である(非特許文献7)。
【0007】
そこで、これらの問題を解決するために、パクリタキセルのDDS化の検討が試みられている。現在開発中のNK105(ナノキャリア)や、既に臨床で使用されているアブラキサンはそれらに該当するものである。
NK105は、親水性のあるポリエチレングリコール(PEG)鎖と、疎水基であるポリアスパラギン酸鎖が鎖状に結合したブロック共重合体にパクリタキセルを混合させることでつくられた高分子ミセル製剤であり、クレモホールELを使用せずにパクリタキセルの溶解度を飛躍的に向上することができるため、溶媒毒性を回避した注射液の提供を可能としている。上記NK105の非臨床試験では、すぐれた血中滞留性と抗腫瘍効果、ならびに神経毒性の軽減が認められ、現在臨床第II相試験が行われている(非特許文献8)。また、同じくパクリタキセルのDDS製剤であるアブラキサンは、ヒト血清アルブミンを添加物とした均一なナノ粒子製剤である。NK105と同様にクレモホールELおよびエタノールを溶媒として使用しないため、溶剤毒性の危険性を免れることができ、かつ前投与を必要とせずに、短時間で高用量のパクリタキセルを投与することができる。(非特許文献9)。このように水難溶性薬物は、DDS化することで溶媒毒性による副作用の危険性を免れることができるため、製剤の安全域を広げることが可能である。さらに、DDS製剤はその特性上、薬剤本来が有する毒性を軽減しながら、抗腫瘍効果を維持、もしくは高めることが期待される。
【0008】
上記KRN5500においても、溶媒毒性による副作用、さらにはKRN5500自体の毒性を軽減しながら、かつ効果を高めることが求められる。したがって、これらの課題を解決するために、これまでKRN5500のミセル化(特許文献3,非特許文献10、非特許文献11)やリポソーム化の検討がなされている。KRN5500のミセル化は、親水性ポリマーセグメントと疎水性ポリマーセグメントを有するブロックコポリマー(PEG−P(C16, BLA))およびKRN5500をそれぞれN,N−ジメチルホルムアミド(DMF)またはジメチルスルホキシド(DMSO)に溶解し、それらを混合攪拌した後、透析、超音波処理を行うことで得られる(非特許文献12)。このKRN5500ミセルの毒性ならびに抗腫瘍効果を評価しており、毒性に関しては、ラットにおけるブレオマイシン(BLM)モデルによる肺毒性において単体と比べて軽減されることが報告されている。一方、抗腫瘍効果に関しては、ヌードマウスにヒト胃癌細胞株MKN-45細胞を移植した系において、KRN5500単体とほぼ同等であり、単体を上回る効果は得られていない。また、これらミセルの粒子サイズは、散乱強度によると2つのピークが検出されており、粒子径の制御が困難である。また、粒度分布も81nm〜390nmと広く、実用可能な注射液として好ましくない。さらに、有機溶媒を用いている点や透析、超音波処理を必要とする製造法を用いている点など、スケールアップに適した製造法ではない。
このようにKRN5500ミセルは、薬効面や製造法、粒子サイズなど多くの課題が残されており、臨床で耐えられるレベルまでに至っていないのが現状である。したがって、KRN5500の有効性を得るためには、有機溶媒やクレモホールELを用いずに可溶化することができ、かつKRN5500本来の毒性および溶剤毒性による副作用を軽減できる製剤の開発が望まれる。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】特開昭59−161396号公報
【特許文献2】特開平5−186494号公報
【特許文献3】特開平11−335267号公報
【非特許文献】
【0010】
【非特許文献1】Kamishouhara M.ら、 J. Antibiotics 48 1995
【非特許文献2】Oncology Research,6,383-390,1994
【非特許文献3】Jpn J Cancer Chemother,1997,24,1571-1577
【非特許文献4】Cancer Chemother Pharm.,2001,47,404-410
【非特許文献5】Clinical Cancer Research 2003 9 5178-5186
【非特許文献6】Jpn J Clin Oncol,2003 6 302-308
【非特許文献7】Mitchell J.B.ら、Lancet. 342, 1993, 1428
【非特許文献8】Br J Cancer 92, 2005, 1240-1246
【非特許文献9】J Clin Oncol. 23, 2005, 7794-7803
【非特許文献10】Jpn.J Cancer Res.90、1999、122-128
【非特許文献11】Jpn.J Cancer Res., 93,2002,1237-1243
【非特許文献12】J Control.Release,55,1998,219-229
【発明の概要】
【発明が解決しようとする課題】
【0011】
水難溶性スピカマイシン誘導体(KRN5500)を、有機溶媒またはクレモホールELを使わずに可溶化することができ、これにより溶媒毒性による副作用の危険性から免れ、また、投与後血中で長時間薬物濃度を維持することができるとともに、KRN5500自体の毒性を軽減しながら効果を維持または高めることができるKRN5500製剤を提供することを目的としている。また、このKRN5500製剤は、バイヤル中での安定性にも非常に優れていることが望まれる。
【課題を解決するための手段】
【0012】
上記課題は以下の本発明により解決される。
本発明者は、上記目的を達成すべく検討したところ、リポソームの膜構成成分であるリン脂質およびコレステロールの脂質組成比をコントロールすることにより、水難溶性スピカマイシン誘導体(KRN5500)をリポソーム化できることを見出した。また、このKRN5500リポソーム製剤の抗腫瘍効果について検討するうちに、アシル鎖長の短いリン脂質(ミリストイル、C14)を用いると抗腫瘍効果を高めることができることを見出した(ただし、この場合、脂質膜構成成分としてコレステロールを約半量含む)。KRN5500は脂溶性薬物であるため、その特性上、脂質膜に内封されると考えられるが、リン脂質のアシル鎖長を短くすることにより、アシル鎖長とKRN5500の脂肪酸側鎖との疎水性相互作用がやや弱まり、その結果、血中では長時間薬物濃度を維持しながらも、患部において効率良くKRN5500を放出させることができるためだと考えられる。また、膜構成成分としてコレステロールを含まない膜処方においても同様に抗腫瘍効果が高まることを見出した。これらの結果より、高い抗腫瘍効果を得るためには、血中での安定性と患部におけるKRN5500の放出性のバランスが非常に重要であることが分かるが、これを支配しているのが膜構成成分であるリン脂質のアシル鎖長とKRN5500の脂肪酸側鎖の疎水性相互作用だと推測される。この疎水性相互作用が強すぎると脂質膜の流動性が抑制されるため、患部でのKRN5500の放出性が抑制され、その結果、期待される抗腫瘍効果は得られないと推測される。一方、この疎水性相互作用が弱すぎると、血中での安定性が低下し直ちに消失するため、同様に期待される抗腫瘍効果は得られない。したがって、KRN5500を血中では安定に保持しながら、患部では効率良く放出できるリポソーム製剤が求められ、膜の流動性が高いほど(ただしコレステロールを含む場合)上記目的を達成できうることが明らかとなった。これらから、上記課題を解決するものとして、以下の本発明を提供する。
【0013】
(1)コレステロールが0〜50mol%および飽和脂肪酸のアシル鎖の鎖長がC14〜C18のいずれかであるリン脂質100〜50mol%を含有するリポソームに、化学式(I)で示されるスピカマイシン誘導体を有するリポソーム製剤:
【化3】
(式中、R1=H、R2=OHであって、Rは炭素数9〜15の直鎖のもしくは分岐したアルキルまたは炭素数10〜17の直鎖のアルケニルである。)。
(2)リポソーム製剤のリポソーム膜が、以下の1)〜3)のいずれかである(1)に記載のリポソーム製剤:
1)リポソーム膜が、アシル鎖の鎖長が18である飽和脂肪酸を有するリン脂質を主構成成分として有する、アシル鎖部分がジステアロイルまたはパルミトイルステアロイルまたはステアロイルパルミトイルまたはミリストイルステアロイルまたはステアロイルミリストイルであるのが好ましい。
2)リポソーム膜が、アシル鎖の鎖長が16である飽和脂肪酸を有するリン脂質およびコレステロールを主構成成分として有し、両成分のモル比が80:20〜50:50である、アシル鎖部分がジパルミトイルまたはパルミトイルミリストイルまたはミリストイルパルミトイルであるのが好ましい。
3)リポソーム膜が、アシル鎖の鎖長が14である飽和脂肪酸を有するリン脂質およびコレステロールを主構成成分として有し、両成分のモル比が60:40〜50:50である、アシル鎖部分がジミリストイルであるのが好ましい。
リポソーム膜の全脂質に対する主構成成分の含有率は50質量%以上、好ましくは80質量%以上であることが望ましい。
(3) 前記リン脂質がフォスファチジルコリンである(1)または(2)に記載のリポソーム製剤。
(4)前記、基R=CH3(CH2)8CH=CHCH=CH-であり、化学式(I)の化合物が(6−[4−deoxy−4−(2E,4E)−tetradecadienoylglycyl]−amino−L−glycero−b−L−mannnoheptopyranosyl)amino−9H−purine)(KRN5500と称される)である(1)〜(3)のいずれかに記載のリポソーム製剤。
(5)前記リポソーム製剤が親水性高分子鎖を有する(1)〜(4)のいずれかに記載のリポソーム製剤。
(6)化学式(I)で示されるスピカマイシン誘導体のAUC(血漿中濃度−時間曲線下面積)が、該スピカマイシン誘導体単体に比べて45倍以上増強することができる(1)〜(5)のいずれかに記載のリポソーム製剤。
【発明の効果】
【0014】
上記のような本発明に係るリポソーム製剤は、水難溶性スピカマイシン誘導体(KRN5500)を安定に保持することができ、膜構成成分であるリン脂質と他の脂質の組成比や、リン脂質のアシル鎖長をコントロールすることで、血中滞留性に優れながらも、患部では効率よくKRN5500を放出することを可能にし、KRN5500本来の抗腫瘍効果をさらに高めることができ、かつ、薬剤の毒性による副作用を軽減することができる。また、上記発明に至る過程で、本発明のKRN5500製剤は、バイヤル中での安定性が良いことが明らかになった。
【図面の簡単な説明】
【0015】
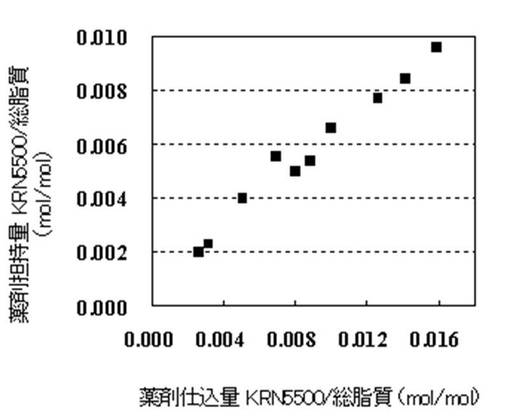
【図1】図1は、薬剤仕込み量に対するリポソームの薬剤担持量を示すグラフである。
【図2】図2は、薬剤仕込み量に対するリポソームへの薬剤封入効率を示すグラフである。
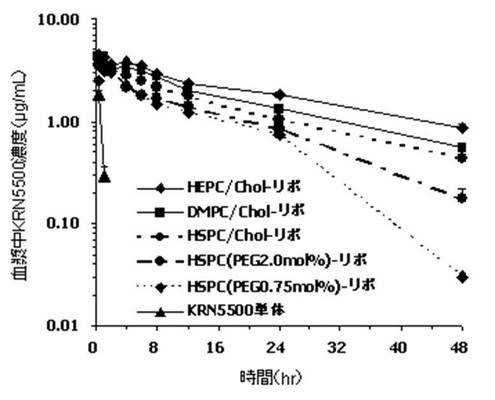
【図3】図3は、リポソーム製剤等を、ラットに投与し、投与後、所定時間経過後の血漿中のKRN5500濃度を吸光度測定によって定量したグラフである。
【図4】図4は、KRN5500リポソーム製剤のin vitroでの殺細胞効果を示すグラフである。
【図5】図5は、マウスにリポソーム製剤等を投与した後の経過日数における腫瘍体積を示すグラフである。
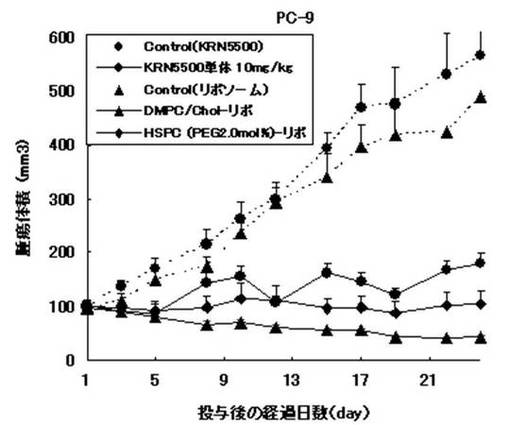
【図6】図6は、マウスにリポソーム製剤等を投与した後の推定腫瘍体積を示すグラフである。
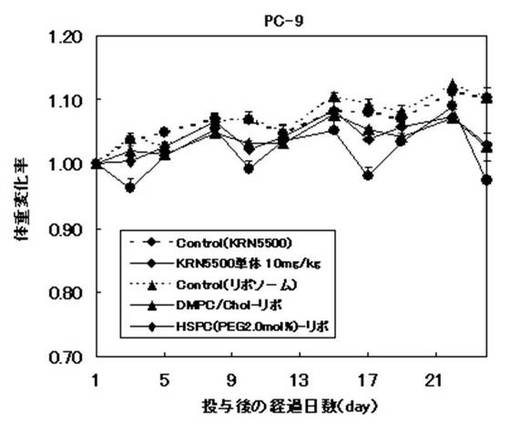
【図7】図7は、マウスにリポソーム製剤等を投与した後のマウスの体重変化率を示すグラフである。
【発明を実施するための形態】
【0016】
以下、本発明をより詳細に説明する。
「水難溶性スピカマイシン誘導体」は、水に不溶もしくは難溶のスピカマイシン骨格を有する化合物の総称である。具体的には、下記式(I)で示され、式中、R1=H、R2=OHであって、Rは炭素数9〜15の直鎖のもしくは分岐したアルキルまたは炭素数10〜17の直鎖のアルケニルを有する誘導体である。好ましくは、R=CH3(CH2)8CH=CHCH=CH-、(6−〔4’-N-(N’-trans, trans-2,4-テトラデカジエノイルグリシル)-スピカミニル−アミノ〕プリン)を有する誘導体であり、その化合物は式(II)で示される(6−[4−deoxy−4−(2E,4E)−tetradecadienoylglycyl]−amino−L−glycero−b−L−mannnoheptopyranosyl)amino−9H−purine)(KRN5500)、あるいは、R=CH3(CH2) 10−、(6−〔4’-N-(N’ -ジデカノイルグリシル)-スピカミニル−アミノ〕プリン)(SPM VIIIまたはSPM Xと称される)であり、より好ましくは(6−[4−deoxy−4−(2E,4E)−tetradecadienoylglycyl]−amino−L−glycero−b−L−mannnoheptopyranosyl)amino−9H−purine)(KRN5500)である。
【0017】
水難溶性スピカマイシン誘導体は、活性体SAN−Gly(4−N−glycylspicamycin aminonucleoside)のプロドラッグである。該プロドラッグの脂肪酸側鎖は癌細胞の細胞膜の透過に必要であり、細胞膜透過時に脂肪酸とグリシンの間で酵素により加水分解され切断されるものと考えられる。そのようにして生じたSAN−Glyが細胞内の蛋白合成系に作用し、癌細胞に対する殺細胞効果を示す。このプロドラッグ誘導体合成の過程でグリシンと脂肪酸部分のアミド結合が重要であり、この部分が化学変換されると抗腫瘍活性が消失することが報告されている。
【0018】
【化4】
【化5】
【0019】
本発明では、上記水難溶性スピカマイシン誘導体(KRN5500)を含有するリポソーム製剤を提供する。
リポソームは、リン脂質二重膜からなり、脂質の疎水性基と親水性基との極性に基づいて生ずる膜により外界から隔てられた空間を形成する構造を有する閉鎖小胞の水性分散液である。膜を隔てて閉鎖小胞内外の水相は、それぞれ内水相、外水相と称される。リポソーム製剤は、リポソームを担体とし、これに薬剤を担持させたものをいう。本発明では、このリポソームに、薬剤として水難溶性スピカマイシン誘導体(KRN5500)を担持させる。
【0020】
上記薬剤を担持するリポソーム膜基材である「リン脂質」は、生体膜の主要構成成分であり、分子内に長鎖アルキル基より構成される疎水性基とリン脂質基より構成される親水性基のグループを持つ両親媒性物質である。リン脂質としては、フォスファチジルコリン(=レシチン)、フォスファチジルグリセロール、フォスファチジン酸、フォスファチジルエタノールアミン、フォスファチジルセリン、フォスファチジルイノシトール、さらにスフィンゴミエリンなどのスフィンゴリン脂質、カルジオリピン等の天然あるいは合成のリン脂質もしくはこれらの誘導体、糖類を結合させた誘導体(糖脂質)およびこれらを常法にしたがって水素添加した水素添加リン脂質などを挙げることができる。これらのうちでも、水素添加大豆フォスファチジルコリン、水素添加卵黄フォスファチジルコリン、ジステアロイルフォスファチジルコリン、ジパルミトイルフォスファチジルコリン、ジミリストイルフォスファチジルコリンが好ましい。またリポソームは、上記主膜材とともに他の膜成分を含んでいてもよい。たとえば、リン脂質以外の脂質もしくはその誘導体(以下、他の脂質類と称することもある)を含む。
「リン脂質以外の脂質」とは、分子内に長鎖アルキル基等より構成される疎水性基を有し、リン酸基を分子内に含まない脂質であり、特に限定されないがグリセロ糖脂質、スフィンゴ糖脂質およびコレステロールなどのステロール類等およびこれらの水素添加物などの誘導体を挙げることができる。コレステロール誘導体とは、シクロペンタノヒドロフェナントレン環を有するステロール類であり、具体例としては特に限定されないがコレステロールが挙げられる。
【0021】
本発明におけるリポソームは、リン脂質およびコレステロールの脂質組成比をコントロールすることによって製剤化することができ、また、用いるリン脂質のアシル鎖長を短くすることによって抗腫瘍効果を高めることができる。
アシル鎖部分がジステアロイル、パルミトイルステアロイル、ステアロイルパルミトイル、ミリストイルステアロイルおよびステアロイルミリストイルからなる群から選択されるリン脂質である場合は、リン脂質100mol%またはリン脂質70〜50mol%、コレステロール30〜50mol%が好ましい。アシル鎖部分がジパルミトイル、パルミトイルミリストイルおよびミリストイルパルミトイルからなる群から選択されるリン脂質である場合は、リン脂質80〜50mol%、コレステロール20〜50mol%が好ましい。リン脂質70〜50mol%、コレステロール30〜50mol%がより好ましい。アシル鎖部分がジミリストイルであるリン脂質である場合は、リン脂質50〜60mol%、コレステロール50〜40mol%が好ましい。これらの範囲であれば薬剤担持量を下げることを防ぎつつ膜を安定化させてスピカマイシン誘導体をリポソーム製剤化できる。
【0022】
また、膜を構成する脂質の物性を変化させ、膜に所望の特性を付与するため、膜が修飾されてもよい。具体的には、脂質膜に親和性のある疎水性本体に修飾基が連結された誘導体を膜中に含むことができ、疎水性本体は、通常、脂質である。この脂質部分は、リン脂質またはリン脂質以外の脂質のいずれか、またはどちらであってもよく特に限定されない。たとえばリン脂質、長鎖脂肪族アルコール、ステロール、ポリオキシプロピレンアルキル、またはグリセリン脂肪酸エステル等が挙げられる。
修飾基としては、特に限定されないが荷電性基、水溶性多糖類などの親水性基、親水性高分子鎖等が挙げられ、これらの1種または2種以上の組み合わせでもよい。
荷電性基は、特に限定されないが、アミノ基、アミジノ基、グアジニノ基などの塩基性官能基、酸性官能基などが挙げられ、これらの基を含む荷電物質で導入することができる。
塩基性官能基を有する荷電物質としては、特開昭61−161246号公報に開示されたDOTMA、特表平5−508626号公報に開示されたDOTAP、特開平2−292246号公報に開示されたトランスフェクタム、特開平4−108391号公報に開示されたTMAG、国際公開第97/42166号・パンフレットに開示された3,5-ジペンタデシロキシベンズアミジン塩酸塩、DOSPA、TfxTM-50、DDAB、DC-CHOL、DMRIEなどが挙げられる。
酸性官能基を有する荷電物質としては、ガングリオシドGM1、ガングリオシドGM3等のシアル酸を有するガングリオシド類、N-アシル-L-グルタミン酸等の酸性アミノ酸系界面活性剤などが挙げられる。
上記荷電物質が、脂質に、塩基性官能基を有する化合物が結合した物質である場合には、カチオン化脂質と称される。カチオン化脂質の脂質部分はリポソームの脂質二重膜中に安定化され、塩基性官能基部分は担体の脂質二重層の膜表面上(外膜表面上および/または内膜表面上)に存在することができる。カチオン化脂質で膜を修飾することにより、リポソーム膜と細胞との接着性等を高めることができる。
【0023】
水溶性多糖類としては、特に限定されないが、たとえばグルクロン酸、シアル酸、デキストラン、プルラン、アミロース、アミロペクチン、キトサン、マンナン、シクロデキストリン、ペクチン、カラギーナンなどの水溶性多糖類などが挙げられる。水溶性多糖類の誘導体は、糖脂質などが挙げられる。
上記荷電物質、水溶性多糖類による膜修飾率は、必要に応じて適宜設定することができる。
親水性高分子としては、特に限定されないがポリエチレングリコール、フイコール、ポリビニルアルコール、スチレン−無水マレイン酸交互共重合体、ジビニルエーテル−無水マレイン酸交互共重合体、ポリビニルピロリドン、ポリビニルメチルエーテル、ポリビニルメチルオキサゾリン、ポリエチルオキサゾリン、ポリヒドロキシプロピルオキサゾリン、ポリヒドロキシプロピルメタアクリルアミド、ポリメタアクリルアミド、ポリジメチルアクリルアミド、ポリヒドロキシプロピルメタアクリレート、ポリヒドロキシエチルアクリレート、ヒドロキシメチルセルロース、ヒドロキシエチルセルロース、ポリアスパルトアミド、合成ポリアミノ酸などが挙げられる。
親水性高分子は、リポソーム膜を修飾するための構造を有していることが好ましい。特に、親水性高分子鎖の一端に該構造を有していることが好ましい。すなわち、修飾に使用される親水性高分子は、親水性高分子の本体部分とリポソーム膜を修飾するための構造部分とからなっていることが好ましい。該構造が脂質等の疎水性部分である場合は、該疎水性部分がリポソーム膜に挿入されるかたちで親水性高分子の本体部分がリポソーム外表面上から突出するように固定化され、該構造がリポソーム膜構成成分と共有結合しうる反応性官能基である場合は、リポソームの外表面に露出しているリン脂質等のリポソーム膜構成成分と共有結合することにより親水性高分子の本体部分がリポソーム外表面上から突出するように固定化される。
【0024】
次に、親水性高分子本体と結合して親水性高分子−疎水性高分子化合物を形成せしめるために使用される疎水性化合物について以下に説明する。
当該疎水性化合物は、特に限定されない。例えば、疎水性の領域を有する化合物(疎水性化合物)を挙げることができる。疎水性化合物としては、例えば、リン脂質やステロール等の他の脂質類、あるいは、長鎖脂肪族アルコール、グリセリン脂肪酸エステル等が挙げられる。中でも、リン脂質が好ましい態様の一つである。また、これらの疎水性化合物は反応性官能基を有してもよい。反応性官能基によって形成される結合としては共有結合が望ましく、具体的にはアミド結合、エステル結合、エーテル結合、スルフィド結合、ジスルフィド結合、などが挙げられるが特に限定されない。
上記リン脂質に含まれるアシル鎖は、飽和脂肪酸であることが望ましい。アシル鎖の鎖長は、C14〜C20が望ましく、さらにはC14〜C18であることが望ましい。アシル鎖としては、例えば、ジミリストイル、ジパルミトイル、ジステアロイル、パルミトイルステアロイルが挙げられる。
リン脂質は、特に制限されない。リン脂質としては、例えば、上記親水性高分子と反応可能な官能基を有するものを使用することができる。このような親水性高分子と反応可能な官能基を有するリン脂質の具体例としては、アミノ基を有するフォスファチジルエタノールアミン、ヒドロキシ基を有するフォスファチジルグリセロール、カルボキシ基を有するフォスファチジルセリンが挙げられる。上記のフォスファチジルエタノールアミンを使用するのが好適な様態の1つである。
親水性高分子の脂質誘導体は、上記の親水性高分子と上記の脂質とからなる。上記の親水性高分子と上記の脂質との組み合わせは、特に限定されない。目的に応じて適宜組み合わせたものを使用することができる。例えば、リン脂質、ステロール等の他の脂質類、長鎖脂肪族アルコール、グリセリン脂肪酸エステルの中から選ばれる少なくとも1つと、PEG、PG、PPGの中から選ばれる少なくとも1つとが結合した親水性高分子の誘導体が挙げられる。具体的には、ポリオキシプロピレンアルキルなどが挙げられ、特に、親水性高分子がポリエチレングリコール(PEG)である場合において脂質としてリン脂質、コレステロールを選択するのが好適な態様のひとつである。このような組み合わせによるPEGの脂質誘導体としては、例えば、PEGのリン脂質誘導体またはPEGのコレステロール誘導体が挙げられる。
【0025】
親水性高分子の脂質誘導体は、脂質の選択により、正電荷、負電荷、中性の選択が可能である。例えば、脂質としてDSPEを選択した場合、リン酸基の影響で負電荷を示す脂質誘導体となり、また脂質としてコレステロールを選択した場合、中性の脂質誘導体となる。脂質は、その目的に応じ、選択することが可能である。
PEGの分子量については特に限定されないが、通常、500〜10,000ダルトン、好ましくは1,000〜7,000ダルトン、より好ましくは2,000〜5,000ダルトンである。
PGの分子量についてはとくに限定されないが、通常100〜10,000ダルトンであり、好ましくは200〜7,000ダルトン、より好ましくは400〜5,000ダルトンである。
PPGの分子量については特に限定されないが、通常100〜10,000ダルトンであり、好ましくは200〜7,000ダルトン、より好ましくは10,000〜5,000ダルトンである。
これらの中でも、PEGのリン脂質誘導体が好ましい態様の一つとして挙げられる。PEGのリン脂質誘導体としては、例えば、ポリエチレングリコール−ジステアロイルフォスファチジルエタノールアミン(PEG−DSPE)が挙げられる。PEG−DSPEは、汎用の化合物であり入手容易であることから好ましい。
【0026】
このような親水性高分子の脂質誘導体を用いて表面修飾されたリポソームは、血漿中のオプソニン蛋白質等が当該リポソームの表面へ吸着するのを防止して当該リポソームの血中安定性を高め、肝臓、脾臓等の細網内皮系組織(RES)での捕捉を回避することが可能となり、標的とする組織や細胞への送達性を高めることができる。すなわち、高い血中滞留性が得られる。これにより、腫瘍組織の血管透過性が亢進した組織に受動的に集積させることが可能となる。
なお本発明において、「血中滞留性」とは、薬剤が担持されたリポソーム製剤を投与した宿主において、該リポソーム製剤に担持された薬剤が血液中に存在する性質を意味する。薬剤がリポソームから放出されると速やかに血中から消失し、薬剤が暴露した部位へ作用する。血中滞留性が良いと、より少ない量の薬剤を投与することが可能となる。
上記親水性高分子脂質誘導体による膜脂質(総脂質)修飾率は、膜脂質に対する比率で、通常0.1〜20mol%、好ましくは0.1〜10mol%、より好ましくは0.5〜10mol%とすることができる。さらには0.5〜4mol%である。
なお本発明において、「総脂質」とは、膜を構成する脂質のうちから親水性高分子脂質誘導体を除いた総脂質をいう。
【0027】
本発明において、上記のようなリポソームへの水難溶性スピカマイシン誘導体の担持量、すなわちリポソーム製剤の薬剤/脂質mol比は、水難溶性スピカマイシン誘導体がリポソーム膜に含有されることから、リポソームの安定性および封入効率の観点上、通常、0.1以下であり、好ましくは0.001〜0.03である。
このリポソームへの薬剤担持あるいはリポソーム製剤の薬剤含有とは、リポソーム製剤(分散液)中、薬剤がリポソームに担持され、保持されて存在する状態を意味し、リポソームに保持されていない薬剤、すなわち分散液の外水相部分には、リポソームとは関係なく自由に存在する薬剤は本質的に含まれないことを意味する。水難溶性スピカマイシン誘導体を薬剤とする本発明のリポソーム製剤では、実質的に、薬剤の少なくとも一部が脂質膜内に含まれるなどして膜に担持されていると考えられるが、本発明のリポソーム製剤は、外水相部分に自由な薬剤が存在しなければよく、薬剤の担持状態は限定されず、リポソーム膜に担持および・または内水相に内包されていてよい。
【0028】
本発明のリポソーム製剤は、投与経路次第で医薬的に許容される安定剤および/または酸化防止剤をさらに含むものであってもよい。これらは製薬学的に添加可能な助剤と総称する。安定化剤としては、特に限定されないがグリセロールまたはスクロースなどの糖類が挙げられる。酸化防止剤としては、特に限定されないがアスコルビン酸、尿酸あるいはトコフェノール同属体例えばビタミンEなどが挙げられる。トコフェノールには、α、β、γ、δの4個の異性体が存在するが本発明においてはいずれも使用できる。
本発明のリポソーム製剤は、投与経路次第で医薬的に許容される添加物をさらに含むものであってもよい。このような添加物の例として、水、生理食塩水、医薬的に許容される有機溶媒、コラーゲン、ポリビニルアルコール、ポリビニルピロリドン、カルボキシビニルポリマー、カルビキシメチルセルロースナトリウム、ポリアクリル酸ナトリウム、アルギン酸ナトリウム、水溶性デキストラン、カルボキシメチルスターチナトリウム、ペクチン、メチルセルロース、エチルセルロース、キサンタンガム、アラビアゴム、カゼイン、ゼラチン、寒天、ジグリセリン、プロピレングリコール、ポリエチレングリコール、ワセリン、パラフィン、ステアリルアルコール、ヒト血清アルブミン(HSA)、マンニトール、ソルビトール、ラクトース、PBS、生体内分解性ポリマー、無血清培地、医薬添加物として許容され、かつリポソーム製剤の安定性に影響しない濃度の界面活性剤、安定性に影響しない濃度とは、全製剤中の15質量%以下をいう。あるいは生体内で許容し得る生理的pHの緩衝液などが挙げられる。使用される添加物は、剤型に応じて上記の中から適宜あるいは組み合わせて選択されるが、これらに限定されるものではない。
【0029】
本発明では、これらの添加物(製薬学的に添加可能な助剤)を含む態様のリポソーム製剤を、医薬組成物として供することができる。本発明の医薬組成物は、室温(一般的に21℃〜25℃)、好ましくは0〜8℃での冷蔵で保存することができる。
上記のようなリポソーム製剤は、薬剤を安定的に担持することができる範囲において、公知のリポソーム製剤化方法を広く用いることができる。
本発明においてリポソーム懸濁液の調製方法としては、水和法(Bangham法)、超音波処理法、逆相蒸発法(Reverse phase evaporation vesicles)、加温法、脂質溶解法、DRV法(Dehydrated/Rehydrated Vesicles)、凍結融解法、エタノール注入法、薄膜法、エクストリュージョン法、高圧吐出型乳化機による高圧乳化法(「ライフサイエンスにおけるリポソーム」寺田、吉村ら編;シュプリンガー・フェアラーク東京(1992))などの各種の公知の技術を採用することができる。
また近年開発された技術として、超高圧による圧縮からの速度変換を利用し、液相下でのジェット流により剪断乳化を行うジェット流乳化法、超臨界二酸化炭素を利用したリポソーム調製技術、後段の整粒工程を簡便化する改良型エタノール注入法などもある。
本発明において、典型的なリポソームの調製工程は、薬剤を封入したリポソームを生成させてリポソーム粗懸濁液を得る工程(i)、リポソーム粗懸濁液の整粒化工程(ii)および外液置換(未封入薬物除去)してリポソーム懸濁液を得る工程(iv) を含み、好ましくは工程(ii)と(iv)の間にさらに親水性高分子による表面修飾工程(iii) を含む。
【0030】
これら工程を行うにあたり、上記に示すような調製方法を必要に応じて適宜に採用することができる。また1方法だけでなく、2以上の方法を選択することもでき、同じまたは別の方法を重複ないし追加することもできる。
一般的にリポソームは相転移点をもつため、リポソームの調製のための外液置換工程(未封入薬物除去工程)(iii) の前段各工程を主膜材の相転移点以上の温度で実施することが好ましい。
【0031】
リポソームの脂質二重膜構造は、ユニラメラ小胞(Small Unilamellar Vesicle, SUV, Large Unilamellar Vesicle, LUV)および複数枚からなる多重ラメラ小胞(Multilamellar Vesicle, MLV)などの膜構造が知られている。
本発明に係るリポソームは、どの膜構造でもよいが、封入効率の点から多重ラメラ小胞のリポソームが好ましい。
整粒化工程をたとえば、上記膜乳化法により行う場合には、エクストルーダーを用いて、市販されているポリカーボネート製などのメンブランフィルターを複数回強制通過させることによりユニラメラ化することができ、粒子径をコントロールすることができる。たとえば、粒子径100nmに整粒する場合には、通常、400nm、200nm、100nmなどのメンブレンフィルターを組み合わせて段階的に整粒することができる。
整粒化工程において、リポソーム粗懸濁液の温度が上記主膜材の相転移点以上であれば、粒子径制御が容易である。
整粒化後のリポソームの大きさは特に限定されないが、球状またはそれに近い形態をとることができ、その粒子径(粒子外径の直径)は特に限定されないが、通常、0.02〜2μm、好ましくは0.03〜0.4μm、より好ましくは0.05〜0.25μmである。この粒子径は、Zetasizer(Malvern Instruments. 3000HS、Zatasizer Nano ZS90)を用いて動的光散乱法により全粒子の直径平均値として測定される。
【0032】
本発明のリポソーム製剤の治療の対象となる腫瘍としては、特に限定されないが固形腫瘍であり、具体的には食道癌、胃癌、大腸癌、結腸癌、直腸癌、膵臓癌、肝臓癌、喉頭癌、肺癌、前立腺癌、膀胱癌、乳癌、子宮癌または卵巣癌が挙げられる。標的部位は、腫瘍の細胞、組織、器官または臓器およびそれらの内部などである。したがって、本発明において、疾患とは前記の腫瘍を意味し、薬剤はそれらに対し抗腫瘍効果を示すことが期待される。
本発明において、「暴露」とは、リポソームの外部へ放出された薬剤が外部環境へ作用を及ぼすことを意味する。具体的には、放出されたスピカマイシン誘導体は標的部位に近接し、癌細胞の細胞膜を透過する時に脂肪酸側鎖が脂肪酸とグリシンの間で酵素的に加水分解され、活性体であるSAN−Glyを生じ、このSAN−Glyが細胞内の蛋白合成系に作用し、抗腫瘍効果を発揮する。このような効果を示すために、リポソーム製剤からの薬剤の放出とリポソーム製剤の血中滞留性との均衡を保つ必要がある。
本発明において、「放出」とは、リポソーム製剤に含まれる薬剤がリポソームから離脱することを意味する。
本発明のリポソーム製剤中に含まれる薬剤は、血漿中において、長時間高濃度で標的部位に暴露することで強い抗腫瘍活性を示すことから放出を制御することが重要となる。
【0033】
本発明のリポソーム製剤は、血漿中の薬剤濃度を高濃度で維持することができる。従来のリポソーム製剤は、血液中から速やかに消失するため、標的部位での暴露時間が短く、充分な効果を期待することは困難である。また、血液中からの速やかな消失は代謝器官である肝臓、脾臓などの臓器に対して薬剤を高濃度で暴露することになり、当該部位での副作用につながるため好ましくない。本発明のリポソーム製剤は、血漿中の薬剤濃度を高濃度で維持することができるため、肝臓、脾臓などの臓器に対する薬剤の暴露を軽減することができ、それに伴い、標的部位において薬剤を長時間暴露することができ、副作用を軽減することができることから好適である。
本発明において、投与後1時間における血漿中の薬剤濃度は、初期値の10%以上であり、好ましくは15%以上である。この「初期値」とは、本発明のリポソーム製剤を投与した直後の理論濃度であり、一般的に、宿主の体重から算出した全血漿量を用いて、投与液量で希釈されたと仮定して算出される。また、各時点の血漿中濃度は、初期値に対する比率として示すことができ、一般的に「% dose」として表記される。
本発明では、薬剤が所望の標的部位に長時間暴露するために使用される。したがって、本発明において、宿主の疾患の予防および/または治療のため、有効量の薬剤を担持するリポソーム製剤を宿主に投与することにより、宿主内で有効量の薬剤を放出し、標的部位に長時間高濃度で暴露するために、宿主(患者)に非経口的に全身あるいは局所的に投与することができる。投与対象の宿主としては、哺乳動物、好ましくはヒト、サル、ネズミ、家畜等が挙げられる。
【0034】
非経口的投与の経路としては、例えば点滴などの静脈注射(静注)、筋肉内注射、腹腔内注射、皮下注射を選択することができ、患者の年齢、症状により適宜投与方法を選択することができる。本発明の担体は、病気に既に悩まされる患者に、疾患の症状を治癒するか、あるいは少なくとも部分的に阻止するために十分な量で投与される。例えば、担体に封入される薬剤の有効投与量は、一日につき体重1kgあたり0.01mgから100mgの範囲で選ばれる。しかしながら、本発明の担体はこれらの投与量に制限されるものではない。投与時期は、疾患が生じてから投与してもよいし、あるいは疾患の発症が予測される時に発症時の症状緩和のために予防的に投与してもよい。また、投与期間は、患者の年齢、症状により適宜選択することができる。
具体的な投与方法としては、リポソーム製剤をシリンジや点滴によって投与することができる。また、カテーテルを患者または宿主の体内、例えば管腔内、例えば血管内に挿入して、その先端を標的部位付近に導き、当該カテーテルを通して、所望の標的部位またはその近傍あるいは標的部位への血流が期待される部位から投与することも可能である。
【実施例】
【0035】
次に実施例を挙げて本発明をさらに詳しく説明するが、本発明はこれら実施例に限定されるべきものではない。
各例で調製された薬剤封入リポソームの各濃度および粒子径は、以下のように求めた。
・ リン脂質濃度(mg/mL):高速液体クロマトグラフィーを用いて定量されるリポソーム懸濁液中でのリン脂質濃度。
・ コレステロール濃度(mg/mL):高速液体クロマトグラフィーを用いて定量されるリポソーム懸濁液中でのコレステロール濃度。
・ 総脂質濃度(mol/L):上記リン脂質濃度およびコレステロール濃度から算出される膜構成成分である脂質の合計モル濃度(mM)。この総脂質中には、PEGを導入するためのPEG誘導体中の脂質(例では、PEG−DSPE中のDSPE)は含まない。
・ 薬剤濃度(mg/mL):上記で得られた製剤をRO水(逆浸透膜浄水)でリン脂質濃度が約20mg/mLとなるように希釈した後、さらにメタノールで20倍希釈した溶液について、264nmでの吸光度を、紫外吸光光度計を用いて高速液体クロマトグラフィーにて定量した。内封されたKRN5500濃度を薬剤量(mg)/製剤全量(mL)で示す。
高速液体クロマトグラフィー試験条件:
カラム:内径6mm、長さ15cmのステンレス管に全多孔性球状シリカゲルを充填
(ナカライテスク(株)COSMOSIL 5C18−ARII)。
カラム温度:30℃付近
移動相:pH6.0のクエン酸緩衝液100mLに液体クロマトグラム用メタノール400mLを加え混合する。
流量:1.5mL/min
・ 薬剤担持量(薬剤/総脂質のモル比):リポソームに内封されたKRN5500濃度を上記総脂質濃度に対する上記薬剤濃度の比から、薬剤/総脂質のモル比で示す。
・ 粒子径(nm):リポソーム分散液20μLを生理食塩水3mLに希釈し、Zatasizer 3000HS(Malvern Instruments)またはZatasizer Nano ZS90で測定した平均粒子径。
以下に使用した各成分の略称および分子量を示す。
DSPC:(ジステアロイルフォスファチジルコリン、分子量790.2、日油)
HSPC:水素添加大豆フォスファチジルコリン(分子量790、リポイド(Lipoid)社製SPC3)
HEPC:水素卵黄大豆フォスファチジルコリン(分子量777、日油)
DPPC:(ジパルミトイルフォスファチジルコリン、分子量734.0、日油)
DMPC:(ジミリストイルフォスファチジルコリン分子量677.9、日油)
Chol:コレステロール(分子量388.66、Solvay)
PEG5000−DSPE:ポリエチレングリコール(分子量5000)−ジステアロイルフォスファチジルエタノールアミン(分子量6081、日油)
KRN5500(分子量589.7、協和発酵キリン(株))
【0036】
[実施例1]
(調製例1〜6)KRN5500リポソーム製剤の調製
KRN5500封入リポソーム製剤を達成しうる膜処方を確認するために、アシル鎖長の異なるリン脂質を用いてリポソーム化を試みた。また、異なるリン脂質/Chol比を有する膜処方についても試みた。調製例1、5、6は100mLスケールで調製し、調製例2、3,4は10mLスケールで製造した。なお、下記は100mLスケールでの調製法を示す。
(1)KRN5500リポソーム生成
各種類のリン脂質、コレステロール、KRN5500を表1に示す所定のモル比となるように秤量し、無水エタノール10mLを添加し、加温溶解した。
得られた脂質/薬剤混合エタノール溶液10mLに、約70℃に加温した内水相(pH6.5の10mMクエン酸一水和物/0.9%塩化ナトリウム溶液)90mLを添加し、超音波装置にて撹拌して粗リポソーム懸濁液を調製した。この粗リポソーム懸濁液を、約70℃に加温したエクストルーダー(The Extruder T.100、Lipexbiomembranes Inc.)に取り付けたフィルター(孔径0.2μm×3回、0.1μm×10回、Whatman社)を順次通し、リポソーム懸濁液を調製した。
(2)表面修飾
上記リポソーム懸濁液を加温状態で維持したまま、PEG5000−DSPEの水溶液(37.7mg/mL)を、表1に示すPEG導入率となるように直ちに添加し、加温撹拌することで、リポソームの膜表面(外表面)をPEG修飾した。加温終了後のリポソーム懸濁液は、速やかに氷冷した。
なお、上記PEG導入率(mol%)=(PEG5000−DSPE/総脂質)×100
である。ここでの総脂質は、PEG誘導体中の脂質(PEG5000−DSPE中のDSPE)は含まない。
(3)外液置換
上記氷冷したPEG修飾後のリポソーム懸濁液を、外水相溶液(10mMクエン酸一水和物/0.9%塩化ナトリウム溶液(pH6.5))を用いてクロスフローろ過システム(ビバフロー MW100,000)により外液置換を行った。なお、高濃度のKRN5500リポソーム製剤を得るために濃縮を行った(調製例1、5、6)。
(4)ろ過滅菌
外液置換後の上記リポソーム懸濁液を用いてろ過滅菌を行い無菌化し、最終製剤とした。ろ過滅菌後のリポソーム懸濁液を高速液体クロマトグラフィーを用いて、リン脂質、CholおよびKRN5500濃度を定量した。リン脂質濃度、Chol濃度の総和を総脂質濃度とし、封入されたKRN5500量を求めた。
上記で得られたKRN5500リポソーム製剤の膜組成比、薬剤担持量(薬剤濃度、薬剤/総脂質モル比)粒子径を表1に示す。
調製例1〜4に示すように、コレステロールを約半量含む膜処方の薬剤担持量は、リン脂質のアシル鎖長に関わらず、ほぼKRN5500/総脂質量=0.01(mol/mol)を示した。一方、コレステロールを含まない膜処方(調製例5および6)は、脂質/薬剤混合エタノール溶液の調製時において、仕込みのKRN5500量を約2倍に増やすことができるため、得られたリポソーム製剤の薬剤担持量も約2倍に増加した。
また、製剤化における脂質組成比の影響を検討した結果、表2に示すように、リン脂質のアシル鎖長および膜組成比によってリポソーム化が困難な膜処方があることが明らかとなった。リン脂質のアシル鎖長が長い場合は(ジステアロイル、パルミトイルステアロイル)コレステロールを含まなくてもリポソーム化することができるが、短い場合は(ジパルミトイル、ジミリストイル)、コレステロールを含まないとゲル化が生じ、製剤化できないことが明らかとなった。特にジミリストイル(C14)を用いる場合は、リン脂質に対して約半量のコレステロールが必要であることが明らかとなった。これは、コレステロールの膜安定化効果が影響していると考えられる。すなわち、アシル鎖長が長ければ疎水性相互作用が強まるため、コレステロールによる膜安定化効果がなくてもKRN5500を安定に内封することができると推測される。一方、アシル鎖長が短いと疎水性相互作用が弱まり、膜の流動性が大きくなるため、KRN5500を膜に安定に内封することができず、その結果、KRN5500自体の凝集が起こり、ゲル化してしまうと推測される。
【0037】
【表1】
【0038】
【表2】
【0039】
[実施例2]
本発明のリポソームにおいて、最大薬剤封入量を得るために必要な仕込み量を調べた。
(調製例7〜16)
HSPC/Chol(モル比)=54/46膜処方において、仕込みのKRN5500の量を、薬剤/総脂質(mol/mol)比で0.002、0.003、0.005、0.007、0.008、0.009、0.010、0.012、0.014、0.016、0.018、0.02となる量に変えた以外は、調製例1〜6と同様にしてKRN5500リポソーム製剤を得た。得られたKRN5500リポソーム製剤の薬剤/総脂質および粒子径を表3に示す。また、図1は薬剤担持量を、図2は封入効率のグラフを示す。
KRN5500仕込み量を薬剤/総脂質(mol/mol)=0.002から0.016に変えてリポソーム化の検討を行った結果、図1に示すように、仕込みのKRN5500量の増加と供に、封入量の増加が認められた。仕込みのKRN5500量をさらに増やすことが可能であれば、封入量もさらに増やせることが期待されるが、整粒化工程において目詰まりが発生したり、あるいはKRN5500が脂質エタノール溶液に溶解しなくなったりするため、これ以上仕込み量を増やすことは困難である。したがって、該処方における最大薬剤担持量は、総脂質に対して約1mol%であることが明らかとなった。この結果は他のリン脂質を用いても同様であった。また、KRN5500の封入効率は、仕込み量に関わらず、60〜80%の値を示した(図2)。
【0040】
【表3】
【0041】
[試験例1]薬物動態(血中滞留性)
調製例1、2、4、5、6で調製されたKRN5500リポソーム製剤およびKRN5500単体を、KRN5500量として0.2mg/kgでラットに投与した。投与後、0.25、1、2、4、6、8、12、24、48時間経過後に採血し、遠心分離(5,000 rpm、10分、4℃)して血漿を採取した。引き続き、血漿40μLにメタノール200μLを添加し、遠心分離(10,000rpm、10分、4℃)して上清を採取した。採取した溶液について、264nmでの吸光度を、紫外吸光光度計を用いて高速液体クロマトグラフィーにて定量し、各血漿中のKRN5500濃度を求めた。なお、高速液体クロマトグラフィーの試験条件は、リポソームに内封されたKRN5500濃度の条件と同様である。その結果を表4および図3に示す。
血漿中のKRN5500濃度は、KRN5500単体の場合には、投与後急激に減少し、0.25および1時間のみ検出された。一方、リポソーム製剤はいずれも、投与後48時間まで検出され、KRN5500単体に比べて顕著な滞留時間の延長が認められた。「% dose」で表記すると、KRN5500単体は投与1時間後、約5〜6%であるのに対して、リポソーム製剤は70〜80%であった。また、血漿中濃度−時間曲線下面積(AUC)は、リポソーム製剤はKRN5500単体に比べて60〜100倍大きかった。これらの結果より、本発明におけるリポソーム製剤は、いずれの膜処方においても、血漿中KRN5500濃度を長時間高濃度で維持することが可能であることを確認できた。ただし、コレステロールを含まないHSPC100mol%処方の血中滞留性は、コレステロールを含む処方と比べて、やや低下する傾向があった。特にPEG5000−DSPEの修飾率が0.75mol%の場合は、投与後、直ちに分布し、再び血中に現れる血中動態を示した。しかしながら、PEG5000−DSPEの修飾率を2.0mol%に増加することにより、初期に観察された分布が消失し、血中滞留性の改善が認められた。
【0042】
【表4】
【0043】
[試験例2]殺細胞効果
in vitroでの殺細胞効果評価用には、調製例1、2、3、6および下記に示す調製例17のリポソーム製剤を用いた。
(調製例17)
膜処方がDMPC/Chol(モル比)=54/46となるようにDMPC、CholおよびKRN5500を秤量し、調製例1〜6と同様にしてKRN5500リポソーム製剤を得た(100mLスケール)。得られたKRN5500リポソーム製剤の薬剤/総脂質および粒子径を表5に示す。
【0044】
【表5】
【0045】
図4は、調製例1、2、3、6、17にて調製されたKRN5500リポソーム製剤のin vitroでの殺細胞効果を示す。
ヒト大腸癌株Colo205を10%ウシ胎児血清を含むRPMI培地で、2000cells/90マイクロ L/wellとなるように96wellプレートに蒔き、37℃、5%CO2にて5時間培養した。
被験物質は、KRN5500はDMSOを、KRN5500リポソーム製剤はpH6.5の10mMクエン酸/0.9%塩化ナトリウム溶液を媒体とし、最終濃度(KRN5500として0から10000nMまで10濃度)の200倍の希釈系列を調製し、これをRPMI培地にて20倍希釈した被験物質溶液(検体)を調製した。
細胞を蒔いた96wellプレートに、検体を10マイクロ L/well添加し、37℃、5%CO2にて72時間培養した後、Cell Counting Kit(DOJINDO)を10マイクロ L/well添加し、再び37℃、5%CO2にて約20分間培養し、吸光度(450nm、参照波長650nm)を測定した。
細胞増殖抑制作用は、細胞(−)、培地+被験物質の媒体のみのwellの吸光度(A)を100%阻害、細胞(+)、培地+被験物質の媒体のみのwellの吸光度(B)を0%阻害とし、検体による細胞増殖抑制作用は、抑制作用(%)=100×(B−検体吸光度)/(B−A)で算出した。
膜組成がリン脂質/Chol比=54/46(モル比)のKRN5500リポソーム製剤の殺細胞効果は、リン脂質のアシル鎖長が短いほど活性が高かった(図4)。これは、リン脂質のアシル鎖長が短いほど、アシル鎖長とKRN5500の脂肪酸側鎖の疎水性相互作用が弱く、KRN5500がリポソームから放出しやすいためだと考えられる。また、コレステロールを含まないHSPC100mol%処方においても、KRN5500単体と同程度の高い活性が得られた。
【0046】
[試験例3]抗腫瘍効果(単回投与)
ヒト大腸癌細胞(CoL−1)における単回投与での抗腫瘍効果には、調製例1、5、17と下記に示す調製例18のリポソーム製剤を用いた。
(調製例18)
膜処方がHEPC/Chol(モル比)=54/46となるようにHEPC、CholおよびKRN5500を秤量し、調製例1〜6と同様にしてKRN5500リポソーム製剤を得た(100mLスケールで調製)。得られたKRN5500リポソーム製剤の薬剤/総脂質および粒子径を表6に示す。
【0047】
【表6】
【0048】
ヒト大腸癌株CoL−1腫瘍はヌードマウス(BALB/C、nu/nu、♀)の皮下に接種して通常飼育にて植え継いだ。試験用にヌードマウス背側部皮下に腫瘍断片を接種して通常飼育し、腫瘍体積が100mm3前後に達した段階で腫瘍体積が同程度となるようにn=4で群設定を行った(Day1)。
調製例1、5、17、18で調製したKRN5500リポソーム製剤およびKRN5500単体を、1mg/mLとなるように媒体〔KRN5500の媒体(N,N−ジメチルアセトアミド 3.75%、ポリソルベート80 3.0%、2−アミノエタノール 0.45%、生理食塩水92.8%)、リポソーム製剤の媒体(pH6.5の10mMクエン酸/0.9%塩化ナトリウム溶液)〕で調製し、10mg/kgで静脈内投与した。
KRN5500の媒体群およびリポソーム製剤の媒体群を対照群とした〔Control(Liposome)、Control(KRN5500単体)〕。
腫瘍体積は、Day1より2〜3日間隔で腫瘍の長径(Lmm)、短径(Wmm)、厚さ(Hmm)をノギスで測定し、L×W×H/2mm3で算出した。
図5は、投与後の経過日数における腫瘍体積を示す。KRN5500の媒体およびリポソーム製剤の媒体を投与したマウスを対照群とした[Control(Liposome),Control(KRN5500単体))。
ヒト大腸癌に対して、リポソーム製剤およびKRN5500単体は、いずれも対照群と比較して有意な腫瘍増殖抑制効果を示した。また、リポソーム製剤は、KRN5500単体に比べ高い持続的な抗腫瘍効果が得られた。特に、調製例6および17のリポソーム製剤の最大抗腫瘍効果はKRN5500単体と同程度であり、かつKRN5500単体よりも著しい持続効果が認められた。
【0049】
[試験例4]抗腫瘍効果(反復投与)
ヒト肺癌細胞株PC−9を10%ウシ胎児血清を含むRPMI培地にて培養し、PBSにて洗浄後、10%Tripsin−EDTA処理し、RPMI培地にて2回洗浄後、ヌードマウス(BALB/C、nu/nu、♀)の背側部皮下に5×106cells/100マイクロ L/個体となるように接種し通常飼育した。腫瘍体積が100mm3前後に達した段階で腫瘍体積が同程度となるようにn=4で群設定を行った(Day1)。
Day1より7日間隔で計4回(Day1、8、15、22)、調製例6ならびに17で調製したリポソーム製剤およびKRN5500単体をそれぞれの媒体で1mg/mLとなるように調製し、10mg/kgを静脈内投与した。KRN5500の媒体群およびリポソーム製剤の媒体群を対照群とした〔Control(Liposome)、Control(KRN5500単体)〕。
Day1より、2〜3日間隔で試験例3に準じて腫瘍体積を測定し、またマウス体重(g)を、体重測定値(g)−腫瘍体積(mm3)/1000で算出し、Day1の値を1とした体重変化率を指標とした。
投与後1、3、5、8、10、12、15、17、19、22、24日後に推定腫瘍体積およびマウスの体重を求めた。その結果を表7ならびに図6および図7に示す。
なお、相対腫瘍体積(RTV)および腫瘍増殖抑制率(TGIR)は下記の式を用いて算出した。
【0050】
相対腫瘍体積(RTV)=Day Xの腫瘍体積/Day 1の腫瘍体積
腫瘍増殖抑制率(TGIR、(%))=(1−被験物質投与群のRTV/Control群のRTV)×100
ヒト肺癌細胞に対して、リポソーム製剤およびKRN5500単体は、いずれも対照群と比較して有意な腫瘍増殖抑制効果を示した。さらに、リポソーム製剤はいずれの処方もKRN5500単体に比べて高い抗腫瘍効果が得られ、かつ持続的な効果が認められた。特にDMPC/Chol膜処方のリポソーム製剤は、最も高い抗腫瘍効果が得られた。また、リポソーム製剤はマウスの体重にほとんど影響を与えなかったのに対して、KRN5500単体は投与に伴う一過性の体重減少が観察された。これらの結果より、KRN5500をリポソーム化することで、抗腫瘍効果を高めることができ、かつ副作用も軽減できることが明らかとなった。尚、他の処方のリポソーム製剤もマウスの体重変化にはほとんど影響を及ぼさず、副作用を軽減できることが明らかとなった。また、ラット急性毒性試験の結果では、リポソーム化することによりKRN5500単体で認められた急性死を回避することができ、また肝臓の白色化を軽減できることが明らかとなった。
【0051】
【表7】
【技術分野】
【0001】
本発明は、薬剤として水難溶性スピカマイシン誘導体を含有するリポソーム製剤に関するものである。
【背景技術】
【0002】
癌治療に用いられる抗癌剤は、癌細胞に対する高分子合成阻害活性のパターンから、DNA、RNA、蛋白合成阻害剤に分類される。主に、DNA、RNA合成に作用する抗癌剤の開発が主流であり、この場合は骨髄抑制など限られた器官への毒性が用量制限毒性(DLT)となる例が多い。一方、蛋白合成阻害剤は、肝臓をはじめとする生体維持に必要な重要な蛋白を合成する臓器に毒性が認められることが多く、こういった毒性が、薬効の効果が臨床で認められる前に発現するため、臨床で開発中止になる例が多い。このため、蛋白合成阻害剤は、より高い腫瘍細胞と正常細胞との選択毒性が要求される。
【0003】
式(I)で表されるスピカマイシン[式中、R1=H、R2=OHであって、Rは炭素数9〜15の直鎖のもしくは分岐したアルキルまたは炭素数10〜17の直鎖のアルケニルである]は、
【化1】
HL60細胞から分化誘導された抗腫瘍物質であり、蛋白合成阻害剤として見出された(特許文献1参照)。プリンの6位のアミノ基に特異なアミノヘプトース(以後、このアミノヘプトースをスピカミンと略称する)が結合し、このスピカミンの4位のアミノ基にグリシンがアミド結合し、更にこのグリシンのアミノ基に脂肪酸がアミド結合した構造を有している。該スピカマイシンを抗腫瘍剤として臨床的に使用できうる様に、より毒性が低く、より治療係数が高く、成分的に単一化されたスピカマイシン化合物の提供を目的に、脂肪酸側鎖部分の異なる種々のスピカマイシン誘導体の開発が行われてきた。その結果、これらの各種誘導体において脂肪酸部分の構造が最も抗腫瘍活性に寄与することが見出されており、スピカマイシンX(WO 90/15811号公開公報)(特許願PCT/JP90/00781号)ならびにKRN5500(6−[4−deoxy−4−(2E,4E)−tetradecadienoylglycyl]−amino−L−glycero−b−L−mannnoheptopyranosyl)amino−9H−purine)が、その目的に適合しうる抗腫瘍物質であることが報告されている(特許文献2参照)。
【0004】
これらの中でも特に式(II)で表されるKRN5500は、
【化2】
最も大きな治療指数を有するスピカマイシン誘導体であり、その効果は癌細胞の蛋白合成を阻害することで発揮されるため、新しい抗腫瘍メカニズムを有する新規の抗癌剤として開発が試みられた(非特許文献1)。上記KRN5500は、脂肪酸側鎖部分と細胞とのインターラクションが強力であるため、容易にインターナライゼーションが起こり、細胞内に取り込まれる。細胞内に取り込まれると、この脂肪酸側鎖は細胞膜などにある酵素によって、脂肪酸部分のみが加水分解されて活性本体のSAN−Gly(4−N−glycylspicamycin aminonucleoside)が産生される(非特許文献2)。各細胞がもっているKRN5500のSAN−Glyへの変換活性と、KRN5500のその細胞への殺細胞効果は相関することが認められており、正常細胞ではSAN−Glyへの変換活性は低く、KRN5500が強い殺細胞効果を示す癌細胞株では高く、このことから、KRN5500は比較的癌細胞選択性を有していることが報告されている(非特許文献3)。さらに、KRN5500はアドリアマイシン、ビンクリスチン、マイトマイシンC耐性細胞に対しても親株と同程度の殺細胞効果を示し、ほとんどのP-糖蛋白による薬剤排出機構を免れて多剤耐性細胞に対して有効であることも見出されている。
【0005】
このように、KRN5500は蛋白合成阻害という新しい抗腫瘍メカニズムを有し、かつ既存の抗癌剤の耐性を獲得した癌に対しても高い有効性が期待されるため、新規抗癌剤としての期待度が高く、国内、米国ともに臨床試験が行われた(非特許文献4、非特許文献5、非特許文献6)。しかしながら、国内、米国での第I相臨床試験ではそれぞれに重度の副作用として肺障害、肝障害が出現し、これらの副作用が薬剤の効果が臨床で認められる前に発現したため、開発の中断を余儀なくされている。
KRN5500の副作用に付随する課題の一つはその水難溶性にあり、水への溶解度が著しく低いため有機溶媒またはヒマシ油(クレモホールEL)の使用が余儀なくされている。そのため、KRN5500の薬剤自体の毒性に加えて溶媒毒性による副作用が発現する危険性があり、臨床試験で観察された副作用もこれらの毒性が関与していることが報告されている。また、有機溶媒の使用により製剤のpHが10−11と高いアルカリ性となっているため、臨床試験では中心静脈からの投与を余儀なくされており、そのため投与時間も長く、非常に扱いづらいことも報告されている。このようにKRN5500は薬剤自体の毒性の他に、溶解剤による毒性の問題があり、未だ解決すべき多くの課題があるため、臨床上使用されていないのが現状である。
【0006】
一般的に水難溶性薬物は、このような溶剤毒性による副作用を回避するためにDDS化の検討が試みられている。 例えば、水難溶性薬物としてパクリタキセルが挙げられるが、現在、注射液として投与するために溶媒にクレモホールEL(ポリオキシエチレンヒマシ油)とエタノールを用いている。このクレモホールELは過敏反応を引き起こすことが知られており、アナフィラキシー反応を引き起こし、呼吸困難、顔面紅潮、全身性の発疹、胸痛、頻脈、血圧低下、血管浮腫、蕁麻疹などの発現を認めることがある。そのため、この重篤な過敏症を防ぐためにパクリタキセルの投与時にステロイド剤や坑ヒスタミン薬の前投与薬が必要とされ、大変使いづらいのが現状である(非特許文献7)。
【0007】
そこで、これらの問題を解決するために、パクリタキセルのDDS化の検討が試みられている。現在開発中のNK105(ナノキャリア)や、既に臨床で使用されているアブラキサンはそれらに該当するものである。
NK105は、親水性のあるポリエチレングリコール(PEG)鎖と、疎水基であるポリアスパラギン酸鎖が鎖状に結合したブロック共重合体にパクリタキセルを混合させることでつくられた高分子ミセル製剤であり、クレモホールELを使用せずにパクリタキセルの溶解度を飛躍的に向上することができるため、溶媒毒性を回避した注射液の提供を可能としている。上記NK105の非臨床試験では、すぐれた血中滞留性と抗腫瘍効果、ならびに神経毒性の軽減が認められ、現在臨床第II相試験が行われている(非特許文献8)。また、同じくパクリタキセルのDDS製剤であるアブラキサンは、ヒト血清アルブミンを添加物とした均一なナノ粒子製剤である。NK105と同様にクレモホールELおよびエタノールを溶媒として使用しないため、溶剤毒性の危険性を免れることができ、かつ前投与を必要とせずに、短時間で高用量のパクリタキセルを投与することができる。(非特許文献9)。このように水難溶性薬物は、DDS化することで溶媒毒性による副作用の危険性を免れることができるため、製剤の安全域を広げることが可能である。さらに、DDS製剤はその特性上、薬剤本来が有する毒性を軽減しながら、抗腫瘍効果を維持、もしくは高めることが期待される。
【0008】
上記KRN5500においても、溶媒毒性による副作用、さらにはKRN5500自体の毒性を軽減しながら、かつ効果を高めることが求められる。したがって、これらの課題を解決するために、これまでKRN5500のミセル化(特許文献3,非特許文献10、非特許文献11)やリポソーム化の検討がなされている。KRN5500のミセル化は、親水性ポリマーセグメントと疎水性ポリマーセグメントを有するブロックコポリマー(PEG−P(C16, BLA))およびKRN5500をそれぞれN,N−ジメチルホルムアミド(DMF)またはジメチルスルホキシド(DMSO)に溶解し、それらを混合攪拌した後、透析、超音波処理を行うことで得られる(非特許文献12)。このKRN5500ミセルの毒性ならびに抗腫瘍効果を評価しており、毒性に関しては、ラットにおけるブレオマイシン(BLM)モデルによる肺毒性において単体と比べて軽減されることが報告されている。一方、抗腫瘍効果に関しては、ヌードマウスにヒト胃癌細胞株MKN-45細胞を移植した系において、KRN5500単体とほぼ同等であり、単体を上回る効果は得られていない。また、これらミセルの粒子サイズは、散乱強度によると2つのピークが検出されており、粒子径の制御が困難である。また、粒度分布も81nm〜390nmと広く、実用可能な注射液として好ましくない。さらに、有機溶媒を用いている点や透析、超音波処理を必要とする製造法を用いている点など、スケールアップに適した製造法ではない。
このようにKRN5500ミセルは、薬効面や製造法、粒子サイズなど多くの課題が残されており、臨床で耐えられるレベルまでに至っていないのが現状である。したがって、KRN5500の有効性を得るためには、有機溶媒やクレモホールELを用いずに可溶化することができ、かつKRN5500本来の毒性および溶剤毒性による副作用を軽減できる製剤の開発が望まれる。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】特開昭59−161396号公報
【特許文献2】特開平5−186494号公報
【特許文献3】特開平11−335267号公報
【非特許文献】
【0010】
【非特許文献1】Kamishouhara M.ら、 J. Antibiotics 48 1995
【非特許文献2】Oncology Research,6,383-390,1994
【非特許文献3】Jpn J Cancer Chemother,1997,24,1571-1577
【非特許文献4】Cancer Chemother Pharm.,2001,47,404-410
【非特許文献5】Clinical Cancer Research 2003 9 5178-5186
【非特許文献6】Jpn J Clin Oncol,2003 6 302-308
【非特許文献7】Mitchell J.B.ら、Lancet. 342, 1993, 1428
【非特許文献8】Br J Cancer 92, 2005, 1240-1246
【非特許文献9】J Clin Oncol. 23, 2005, 7794-7803
【非特許文献10】Jpn.J Cancer Res.90、1999、122-128
【非特許文献11】Jpn.J Cancer Res., 93,2002,1237-1243
【非特許文献12】J Control.Release,55,1998,219-229
【発明の概要】
【発明が解決しようとする課題】
【0011】
水難溶性スピカマイシン誘導体(KRN5500)を、有機溶媒またはクレモホールELを使わずに可溶化することができ、これにより溶媒毒性による副作用の危険性から免れ、また、投与後血中で長時間薬物濃度を維持することができるとともに、KRN5500自体の毒性を軽減しながら効果を維持または高めることができるKRN5500製剤を提供することを目的としている。また、このKRN5500製剤は、バイヤル中での安定性にも非常に優れていることが望まれる。
【課題を解決するための手段】
【0012】
上記課題は以下の本発明により解決される。
本発明者は、上記目的を達成すべく検討したところ、リポソームの膜構成成分であるリン脂質およびコレステロールの脂質組成比をコントロールすることにより、水難溶性スピカマイシン誘導体(KRN5500)をリポソーム化できることを見出した。また、このKRN5500リポソーム製剤の抗腫瘍効果について検討するうちに、アシル鎖長の短いリン脂質(ミリストイル、C14)を用いると抗腫瘍効果を高めることができることを見出した(ただし、この場合、脂質膜構成成分としてコレステロールを約半量含む)。KRN5500は脂溶性薬物であるため、その特性上、脂質膜に内封されると考えられるが、リン脂質のアシル鎖長を短くすることにより、アシル鎖長とKRN5500の脂肪酸側鎖との疎水性相互作用がやや弱まり、その結果、血中では長時間薬物濃度を維持しながらも、患部において効率良くKRN5500を放出させることができるためだと考えられる。また、膜構成成分としてコレステロールを含まない膜処方においても同様に抗腫瘍効果が高まることを見出した。これらの結果より、高い抗腫瘍効果を得るためには、血中での安定性と患部におけるKRN5500の放出性のバランスが非常に重要であることが分かるが、これを支配しているのが膜構成成分であるリン脂質のアシル鎖長とKRN5500の脂肪酸側鎖の疎水性相互作用だと推測される。この疎水性相互作用が強すぎると脂質膜の流動性が抑制されるため、患部でのKRN5500の放出性が抑制され、その結果、期待される抗腫瘍効果は得られないと推測される。一方、この疎水性相互作用が弱すぎると、血中での安定性が低下し直ちに消失するため、同様に期待される抗腫瘍効果は得られない。したがって、KRN5500を血中では安定に保持しながら、患部では効率良く放出できるリポソーム製剤が求められ、膜の流動性が高いほど(ただしコレステロールを含む場合)上記目的を達成できうることが明らかとなった。これらから、上記課題を解決するものとして、以下の本発明を提供する。
【0013】
(1)コレステロールが0〜50mol%および飽和脂肪酸のアシル鎖の鎖長がC14〜C18のいずれかであるリン脂質100〜50mol%を含有するリポソームに、化学式(I)で示されるスピカマイシン誘導体を有するリポソーム製剤:
【化3】
(式中、R1=H、R2=OHであって、Rは炭素数9〜15の直鎖のもしくは分岐したアルキルまたは炭素数10〜17の直鎖のアルケニルである。)。
(2)リポソーム製剤のリポソーム膜が、以下の1)〜3)のいずれかである(1)に記載のリポソーム製剤:
1)リポソーム膜が、アシル鎖の鎖長が18である飽和脂肪酸を有するリン脂質を主構成成分として有する、アシル鎖部分がジステアロイルまたはパルミトイルステアロイルまたはステアロイルパルミトイルまたはミリストイルステアロイルまたはステアロイルミリストイルであるのが好ましい。
2)リポソーム膜が、アシル鎖の鎖長が16である飽和脂肪酸を有するリン脂質およびコレステロールを主構成成分として有し、両成分のモル比が80:20〜50:50である、アシル鎖部分がジパルミトイルまたはパルミトイルミリストイルまたはミリストイルパルミトイルであるのが好ましい。
3)リポソーム膜が、アシル鎖の鎖長が14である飽和脂肪酸を有するリン脂質およびコレステロールを主構成成分として有し、両成分のモル比が60:40〜50:50である、アシル鎖部分がジミリストイルであるのが好ましい。
リポソーム膜の全脂質に対する主構成成分の含有率は50質量%以上、好ましくは80質量%以上であることが望ましい。
(3) 前記リン脂質がフォスファチジルコリンである(1)または(2)に記載のリポソーム製剤。
(4)前記、基R=CH3(CH2)8CH=CHCH=CH-であり、化学式(I)の化合物が(6−[4−deoxy−4−(2E,4E)−tetradecadienoylglycyl]−amino−L−glycero−b−L−mannnoheptopyranosyl)amino−9H−purine)(KRN5500と称される)である(1)〜(3)のいずれかに記載のリポソーム製剤。
(5)前記リポソーム製剤が親水性高分子鎖を有する(1)〜(4)のいずれかに記載のリポソーム製剤。
(6)化学式(I)で示されるスピカマイシン誘導体のAUC(血漿中濃度−時間曲線下面積)が、該スピカマイシン誘導体単体に比べて45倍以上増強することができる(1)〜(5)のいずれかに記載のリポソーム製剤。
【発明の効果】
【0014】
上記のような本発明に係るリポソーム製剤は、水難溶性スピカマイシン誘導体(KRN5500)を安定に保持することができ、膜構成成分であるリン脂質と他の脂質の組成比や、リン脂質のアシル鎖長をコントロールすることで、血中滞留性に優れながらも、患部では効率よくKRN5500を放出することを可能にし、KRN5500本来の抗腫瘍効果をさらに高めることができ、かつ、薬剤の毒性による副作用を軽減することができる。また、上記発明に至る過程で、本発明のKRN5500製剤は、バイヤル中での安定性が良いことが明らかになった。
【図面の簡単な説明】
【0015】
【図1】図1は、薬剤仕込み量に対するリポソームの薬剤担持量を示すグラフである。
【図2】図2は、薬剤仕込み量に対するリポソームへの薬剤封入効率を示すグラフである。
【図3】図3は、リポソーム製剤等を、ラットに投与し、投与後、所定時間経過後の血漿中のKRN5500濃度を吸光度測定によって定量したグラフである。
【図4】図4は、KRN5500リポソーム製剤のin vitroでの殺細胞効果を示すグラフである。
【図5】図5は、マウスにリポソーム製剤等を投与した後の経過日数における腫瘍体積を示すグラフである。
【図6】図6は、マウスにリポソーム製剤等を投与した後の推定腫瘍体積を示すグラフである。
【図7】図7は、マウスにリポソーム製剤等を投与した後のマウスの体重変化率を示すグラフである。
【発明を実施するための形態】
【0016】
以下、本発明をより詳細に説明する。
「水難溶性スピカマイシン誘導体」は、水に不溶もしくは難溶のスピカマイシン骨格を有する化合物の総称である。具体的には、下記式(I)で示され、式中、R1=H、R2=OHであって、Rは炭素数9〜15の直鎖のもしくは分岐したアルキルまたは炭素数10〜17の直鎖のアルケニルを有する誘導体である。好ましくは、R=CH3(CH2)8CH=CHCH=CH-、(6−〔4’-N-(N’-trans, trans-2,4-テトラデカジエノイルグリシル)-スピカミニル−アミノ〕プリン)を有する誘導体であり、その化合物は式(II)で示される(6−[4−deoxy−4−(2E,4E)−tetradecadienoylglycyl]−amino−L−glycero−b−L−mannnoheptopyranosyl)amino−9H−purine)(KRN5500)、あるいは、R=CH3(CH2) 10−、(6−〔4’-N-(N’ -ジデカノイルグリシル)-スピカミニル−アミノ〕プリン)(SPM VIIIまたはSPM Xと称される)であり、より好ましくは(6−[4−deoxy−4−(2E,4E)−tetradecadienoylglycyl]−amino−L−glycero−b−L−mannnoheptopyranosyl)amino−9H−purine)(KRN5500)である。
【0017】
水難溶性スピカマイシン誘導体は、活性体SAN−Gly(4−N−glycylspicamycin aminonucleoside)のプロドラッグである。該プロドラッグの脂肪酸側鎖は癌細胞の細胞膜の透過に必要であり、細胞膜透過時に脂肪酸とグリシンの間で酵素により加水分解され切断されるものと考えられる。そのようにして生じたSAN−Glyが細胞内の蛋白合成系に作用し、癌細胞に対する殺細胞効果を示す。このプロドラッグ誘導体合成の過程でグリシンと脂肪酸部分のアミド結合が重要であり、この部分が化学変換されると抗腫瘍活性が消失することが報告されている。
【0018】
【化4】
【化5】
【0019】
本発明では、上記水難溶性スピカマイシン誘導体(KRN5500)を含有するリポソーム製剤を提供する。
リポソームは、リン脂質二重膜からなり、脂質の疎水性基と親水性基との極性に基づいて生ずる膜により外界から隔てられた空間を形成する構造を有する閉鎖小胞の水性分散液である。膜を隔てて閉鎖小胞内外の水相は、それぞれ内水相、外水相と称される。リポソーム製剤は、リポソームを担体とし、これに薬剤を担持させたものをいう。本発明では、このリポソームに、薬剤として水難溶性スピカマイシン誘導体(KRN5500)を担持させる。
【0020】
上記薬剤を担持するリポソーム膜基材である「リン脂質」は、生体膜の主要構成成分であり、分子内に長鎖アルキル基より構成される疎水性基とリン脂質基より構成される親水性基のグループを持つ両親媒性物質である。リン脂質としては、フォスファチジルコリン(=レシチン)、フォスファチジルグリセロール、フォスファチジン酸、フォスファチジルエタノールアミン、フォスファチジルセリン、フォスファチジルイノシトール、さらにスフィンゴミエリンなどのスフィンゴリン脂質、カルジオリピン等の天然あるいは合成のリン脂質もしくはこれらの誘導体、糖類を結合させた誘導体(糖脂質)およびこれらを常法にしたがって水素添加した水素添加リン脂質などを挙げることができる。これらのうちでも、水素添加大豆フォスファチジルコリン、水素添加卵黄フォスファチジルコリン、ジステアロイルフォスファチジルコリン、ジパルミトイルフォスファチジルコリン、ジミリストイルフォスファチジルコリンが好ましい。またリポソームは、上記主膜材とともに他の膜成分を含んでいてもよい。たとえば、リン脂質以外の脂質もしくはその誘導体(以下、他の脂質類と称することもある)を含む。
「リン脂質以外の脂質」とは、分子内に長鎖アルキル基等より構成される疎水性基を有し、リン酸基を分子内に含まない脂質であり、特に限定されないがグリセロ糖脂質、スフィンゴ糖脂質およびコレステロールなどのステロール類等およびこれらの水素添加物などの誘導体を挙げることができる。コレステロール誘導体とは、シクロペンタノヒドロフェナントレン環を有するステロール類であり、具体例としては特に限定されないがコレステロールが挙げられる。
【0021】
本発明におけるリポソームは、リン脂質およびコレステロールの脂質組成比をコントロールすることによって製剤化することができ、また、用いるリン脂質のアシル鎖長を短くすることによって抗腫瘍効果を高めることができる。
アシル鎖部分がジステアロイル、パルミトイルステアロイル、ステアロイルパルミトイル、ミリストイルステアロイルおよびステアロイルミリストイルからなる群から選択されるリン脂質である場合は、リン脂質100mol%またはリン脂質70〜50mol%、コレステロール30〜50mol%が好ましい。アシル鎖部分がジパルミトイル、パルミトイルミリストイルおよびミリストイルパルミトイルからなる群から選択されるリン脂質である場合は、リン脂質80〜50mol%、コレステロール20〜50mol%が好ましい。リン脂質70〜50mol%、コレステロール30〜50mol%がより好ましい。アシル鎖部分がジミリストイルであるリン脂質である場合は、リン脂質50〜60mol%、コレステロール50〜40mol%が好ましい。これらの範囲であれば薬剤担持量を下げることを防ぎつつ膜を安定化させてスピカマイシン誘導体をリポソーム製剤化できる。
【0022】
また、膜を構成する脂質の物性を変化させ、膜に所望の特性を付与するため、膜が修飾されてもよい。具体的には、脂質膜に親和性のある疎水性本体に修飾基が連結された誘導体を膜中に含むことができ、疎水性本体は、通常、脂質である。この脂質部分は、リン脂質またはリン脂質以外の脂質のいずれか、またはどちらであってもよく特に限定されない。たとえばリン脂質、長鎖脂肪族アルコール、ステロール、ポリオキシプロピレンアルキル、またはグリセリン脂肪酸エステル等が挙げられる。
修飾基としては、特に限定されないが荷電性基、水溶性多糖類などの親水性基、親水性高分子鎖等が挙げられ、これらの1種または2種以上の組み合わせでもよい。
荷電性基は、特に限定されないが、アミノ基、アミジノ基、グアジニノ基などの塩基性官能基、酸性官能基などが挙げられ、これらの基を含む荷電物質で導入することができる。
塩基性官能基を有する荷電物質としては、特開昭61−161246号公報に開示されたDOTMA、特表平5−508626号公報に開示されたDOTAP、特開平2−292246号公報に開示されたトランスフェクタム、特開平4−108391号公報に開示されたTMAG、国際公開第97/42166号・パンフレットに開示された3,5-ジペンタデシロキシベンズアミジン塩酸塩、DOSPA、TfxTM-50、DDAB、DC-CHOL、DMRIEなどが挙げられる。
酸性官能基を有する荷電物質としては、ガングリオシドGM1、ガングリオシドGM3等のシアル酸を有するガングリオシド類、N-アシル-L-グルタミン酸等の酸性アミノ酸系界面活性剤などが挙げられる。
上記荷電物質が、脂質に、塩基性官能基を有する化合物が結合した物質である場合には、カチオン化脂質と称される。カチオン化脂質の脂質部分はリポソームの脂質二重膜中に安定化され、塩基性官能基部分は担体の脂質二重層の膜表面上(外膜表面上および/または内膜表面上)に存在することができる。カチオン化脂質で膜を修飾することにより、リポソーム膜と細胞との接着性等を高めることができる。
【0023】
水溶性多糖類としては、特に限定されないが、たとえばグルクロン酸、シアル酸、デキストラン、プルラン、アミロース、アミロペクチン、キトサン、マンナン、シクロデキストリン、ペクチン、カラギーナンなどの水溶性多糖類などが挙げられる。水溶性多糖類の誘導体は、糖脂質などが挙げられる。
上記荷電物質、水溶性多糖類による膜修飾率は、必要に応じて適宜設定することができる。
親水性高分子としては、特に限定されないがポリエチレングリコール、フイコール、ポリビニルアルコール、スチレン−無水マレイン酸交互共重合体、ジビニルエーテル−無水マレイン酸交互共重合体、ポリビニルピロリドン、ポリビニルメチルエーテル、ポリビニルメチルオキサゾリン、ポリエチルオキサゾリン、ポリヒドロキシプロピルオキサゾリン、ポリヒドロキシプロピルメタアクリルアミド、ポリメタアクリルアミド、ポリジメチルアクリルアミド、ポリヒドロキシプロピルメタアクリレート、ポリヒドロキシエチルアクリレート、ヒドロキシメチルセルロース、ヒドロキシエチルセルロース、ポリアスパルトアミド、合成ポリアミノ酸などが挙げられる。
親水性高分子は、リポソーム膜を修飾するための構造を有していることが好ましい。特に、親水性高分子鎖の一端に該構造を有していることが好ましい。すなわち、修飾に使用される親水性高分子は、親水性高分子の本体部分とリポソーム膜を修飾するための構造部分とからなっていることが好ましい。該構造が脂質等の疎水性部分である場合は、該疎水性部分がリポソーム膜に挿入されるかたちで親水性高分子の本体部分がリポソーム外表面上から突出するように固定化され、該構造がリポソーム膜構成成分と共有結合しうる反応性官能基である場合は、リポソームの外表面に露出しているリン脂質等のリポソーム膜構成成分と共有結合することにより親水性高分子の本体部分がリポソーム外表面上から突出するように固定化される。
【0024】
次に、親水性高分子本体と結合して親水性高分子−疎水性高分子化合物を形成せしめるために使用される疎水性化合物について以下に説明する。
当該疎水性化合物は、特に限定されない。例えば、疎水性の領域を有する化合物(疎水性化合物)を挙げることができる。疎水性化合物としては、例えば、リン脂質やステロール等の他の脂質類、あるいは、長鎖脂肪族アルコール、グリセリン脂肪酸エステル等が挙げられる。中でも、リン脂質が好ましい態様の一つである。また、これらの疎水性化合物は反応性官能基を有してもよい。反応性官能基によって形成される結合としては共有結合が望ましく、具体的にはアミド結合、エステル結合、エーテル結合、スルフィド結合、ジスルフィド結合、などが挙げられるが特に限定されない。
上記リン脂質に含まれるアシル鎖は、飽和脂肪酸であることが望ましい。アシル鎖の鎖長は、C14〜C20が望ましく、さらにはC14〜C18であることが望ましい。アシル鎖としては、例えば、ジミリストイル、ジパルミトイル、ジステアロイル、パルミトイルステアロイルが挙げられる。
リン脂質は、特に制限されない。リン脂質としては、例えば、上記親水性高分子と反応可能な官能基を有するものを使用することができる。このような親水性高分子と反応可能な官能基を有するリン脂質の具体例としては、アミノ基を有するフォスファチジルエタノールアミン、ヒドロキシ基を有するフォスファチジルグリセロール、カルボキシ基を有するフォスファチジルセリンが挙げられる。上記のフォスファチジルエタノールアミンを使用するのが好適な様態の1つである。
親水性高分子の脂質誘導体は、上記の親水性高分子と上記の脂質とからなる。上記の親水性高分子と上記の脂質との組み合わせは、特に限定されない。目的に応じて適宜組み合わせたものを使用することができる。例えば、リン脂質、ステロール等の他の脂質類、長鎖脂肪族アルコール、グリセリン脂肪酸エステルの中から選ばれる少なくとも1つと、PEG、PG、PPGの中から選ばれる少なくとも1つとが結合した親水性高分子の誘導体が挙げられる。具体的には、ポリオキシプロピレンアルキルなどが挙げられ、特に、親水性高分子がポリエチレングリコール(PEG)である場合において脂質としてリン脂質、コレステロールを選択するのが好適な態様のひとつである。このような組み合わせによるPEGの脂質誘導体としては、例えば、PEGのリン脂質誘導体またはPEGのコレステロール誘導体が挙げられる。
【0025】
親水性高分子の脂質誘導体は、脂質の選択により、正電荷、負電荷、中性の選択が可能である。例えば、脂質としてDSPEを選択した場合、リン酸基の影響で負電荷を示す脂質誘導体となり、また脂質としてコレステロールを選択した場合、中性の脂質誘導体となる。脂質は、その目的に応じ、選択することが可能である。
PEGの分子量については特に限定されないが、通常、500〜10,000ダルトン、好ましくは1,000〜7,000ダルトン、より好ましくは2,000〜5,000ダルトンである。
PGの分子量についてはとくに限定されないが、通常100〜10,000ダルトンであり、好ましくは200〜7,000ダルトン、より好ましくは400〜5,000ダルトンである。
PPGの分子量については特に限定されないが、通常100〜10,000ダルトンであり、好ましくは200〜7,000ダルトン、より好ましくは10,000〜5,000ダルトンである。
これらの中でも、PEGのリン脂質誘導体が好ましい態様の一つとして挙げられる。PEGのリン脂質誘導体としては、例えば、ポリエチレングリコール−ジステアロイルフォスファチジルエタノールアミン(PEG−DSPE)が挙げられる。PEG−DSPEは、汎用の化合物であり入手容易であることから好ましい。
【0026】
このような親水性高分子の脂質誘導体を用いて表面修飾されたリポソームは、血漿中のオプソニン蛋白質等が当該リポソームの表面へ吸着するのを防止して当該リポソームの血中安定性を高め、肝臓、脾臓等の細網内皮系組織(RES)での捕捉を回避することが可能となり、標的とする組織や細胞への送達性を高めることができる。すなわち、高い血中滞留性が得られる。これにより、腫瘍組織の血管透過性が亢進した組織に受動的に集積させることが可能となる。
なお本発明において、「血中滞留性」とは、薬剤が担持されたリポソーム製剤を投与した宿主において、該リポソーム製剤に担持された薬剤が血液中に存在する性質を意味する。薬剤がリポソームから放出されると速やかに血中から消失し、薬剤が暴露した部位へ作用する。血中滞留性が良いと、より少ない量の薬剤を投与することが可能となる。
上記親水性高分子脂質誘導体による膜脂質(総脂質)修飾率は、膜脂質に対する比率で、通常0.1〜20mol%、好ましくは0.1〜10mol%、より好ましくは0.5〜10mol%とすることができる。さらには0.5〜4mol%である。
なお本発明において、「総脂質」とは、膜を構成する脂質のうちから親水性高分子脂質誘導体を除いた総脂質をいう。
【0027】
本発明において、上記のようなリポソームへの水難溶性スピカマイシン誘導体の担持量、すなわちリポソーム製剤の薬剤/脂質mol比は、水難溶性スピカマイシン誘導体がリポソーム膜に含有されることから、リポソームの安定性および封入効率の観点上、通常、0.1以下であり、好ましくは0.001〜0.03である。
このリポソームへの薬剤担持あるいはリポソーム製剤の薬剤含有とは、リポソーム製剤(分散液)中、薬剤がリポソームに担持され、保持されて存在する状態を意味し、リポソームに保持されていない薬剤、すなわち分散液の外水相部分には、リポソームとは関係なく自由に存在する薬剤は本質的に含まれないことを意味する。水難溶性スピカマイシン誘導体を薬剤とする本発明のリポソーム製剤では、実質的に、薬剤の少なくとも一部が脂質膜内に含まれるなどして膜に担持されていると考えられるが、本発明のリポソーム製剤は、外水相部分に自由な薬剤が存在しなければよく、薬剤の担持状態は限定されず、リポソーム膜に担持および・または内水相に内包されていてよい。
【0028】
本発明のリポソーム製剤は、投与経路次第で医薬的に許容される安定剤および/または酸化防止剤をさらに含むものであってもよい。これらは製薬学的に添加可能な助剤と総称する。安定化剤としては、特に限定されないがグリセロールまたはスクロースなどの糖類が挙げられる。酸化防止剤としては、特に限定されないがアスコルビン酸、尿酸あるいはトコフェノール同属体例えばビタミンEなどが挙げられる。トコフェノールには、α、β、γ、δの4個の異性体が存在するが本発明においてはいずれも使用できる。
本発明のリポソーム製剤は、投与経路次第で医薬的に許容される添加物をさらに含むものであってもよい。このような添加物の例として、水、生理食塩水、医薬的に許容される有機溶媒、コラーゲン、ポリビニルアルコール、ポリビニルピロリドン、カルボキシビニルポリマー、カルビキシメチルセルロースナトリウム、ポリアクリル酸ナトリウム、アルギン酸ナトリウム、水溶性デキストラン、カルボキシメチルスターチナトリウム、ペクチン、メチルセルロース、エチルセルロース、キサンタンガム、アラビアゴム、カゼイン、ゼラチン、寒天、ジグリセリン、プロピレングリコール、ポリエチレングリコール、ワセリン、パラフィン、ステアリルアルコール、ヒト血清アルブミン(HSA)、マンニトール、ソルビトール、ラクトース、PBS、生体内分解性ポリマー、無血清培地、医薬添加物として許容され、かつリポソーム製剤の安定性に影響しない濃度の界面活性剤、安定性に影響しない濃度とは、全製剤中の15質量%以下をいう。あるいは生体内で許容し得る生理的pHの緩衝液などが挙げられる。使用される添加物は、剤型に応じて上記の中から適宜あるいは組み合わせて選択されるが、これらに限定されるものではない。
【0029】
本発明では、これらの添加物(製薬学的に添加可能な助剤)を含む態様のリポソーム製剤を、医薬組成物として供することができる。本発明の医薬組成物は、室温(一般的に21℃〜25℃)、好ましくは0〜8℃での冷蔵で保存することができる。
上記のようなリポソーム製剤は、薬剤を安定的に担持することができる範囲において、公知のリポソーム製剤化方法を広く用いることができる。
本発明においてリポソーム懸濁液の調製方法としては、水和法(Bangham法)、超音波処理法、逆相蒸発法(Reverse phase evaporation vesicles)、加温法、脂質溶解法、DRV法(Dehydrated/Rehydrated Vesicles)、凍結融解法、エタノール注入法、薄膜法、エクストリュージョン法、高圧吐出型乳化機による高圧乳化法(「ライフサイエンスにおけるリポソーム」寺田、吉村ら編;シュプリンガー・フェアラーク東京(1992))などの各種の公知の技術を採用することができる。
また近年開発された技術として、超高圧による圧縮からの速度変換を利用し、液相下でのジェット流により剪断乳化を行うジェット流乳化法、超臨界二酸化炭素を利用したリポソーム調製技術、後段の整粒工程を簡便化する改良型エタノール注入法などもある。
本発明において、典型的なリポソームの調製工程は、薬剤を封入したリポソームを生成させてリポソーム粗懸濁液を得る工程(i)、リポソーム粗懸濁液の整粒化工程(ii)および外液置換(未封入薬物除去)してリポソーム懸濁液を得る工程(iv) を含み、好ましくは工程(ii)と(iv)の間にさらに親水性高分子による表面修飾工程(iii) を含む。
【0030】
これら工程を行うにあたり、上記に示すような調製方法を必要に応じて適宜に採用することができる。また1方法だけでなく、2以上の方法を選択することもでき、同じまたは別の方法を重複ないし追加することもできる。
一般的にリポソームは相転移点をもつため、リポソームの調製のための外液置換工程(未封入薬物除去工程)(iii) の前段各工程を主膜材の相転移点以上の温度で実施することが好ましい。
【0031】
リポソームの脂質二重膜構造は、ユニラメラ小胞(Small Unilamellar Vesicle, SUV, Large Unilamellar Vesicle, LUV)および複数枚からなる多重ラメラ小胞(Multilamellar Vesicle, MLV)などの膜構造が知られている。
本発明に係るリポソームは、どの膜構造でもよいが、封入効率の点から多重ラメラ小胞のリポソームが好ましい。
整粒化工程をたとえば、上記膜乳化法により行う場合には、エクストルーダーを用いて、市販されているポリカーボネート製などのメンブランフィルターを複数回強制通過させることによりユニラメラ化することができ、粒子径をコントロールすることができる。たとえば、粒子径100nmに整粒する場合には、通常、400nm、200nm、100nmなどのメンブレンフィルターを組み合わせて段階的に整粒することができる。
整粒化工程において、リポソーム粗懸濁液の温度が上記主膜材の相転移点以上であれば、粒子径制御が容易である。
整粒化後のリポソームの大きさは特に限定されないが、球状またはそれに近い形態をとることができ、その粒子径(粒子外径の直径)は特に限定されないが、通常、0.02〜2μm、好ましくは0.03〜0.4μm、より好ましくは0.05〜0.25μmである。この粒子径は、Zetasizer(Malvern Instruments. 3000HS、Zatasizer Nano ZS90)を用いて動的光散乱法により全粒子の直径平均値として測定される。
【0032】
本発明のリポソーム製剤の治療の対象となる腫瘍としては、特に限定されないが固形腫瘍であり、具体的には食道癌、胃癌、大腸癌、結腸癌、直腸癌、膵臓癌、肝臓癌、喉頭癌、肺癌、前立腺癌、膀胱癌、乳癌、子宮癌または卵巣癌が挙げられる。標的部位は、腫瘍の細胞、組織、器官または臓器およびそれらの内部などである。したがって、本発明において、疾患とは前記の腫瘍を意味し、薬剤はそれらに対し抗腫瘍効果を示すことが期待される。
本発明において、「暴露」とは、リポソームの外部へ放出された薬剤が外部環境へ作用を及ぼすことを意味する。具体的には、放出されたスピカマイシン誘導体は標的部位に近接し、癌細胞の細胞膜を透過する時に脂肪酸側鎖が脂肪酸とグリシンの間で酵素的に加水分解され、活性体であるSAN−Glyを生じ、このSAN−Glyが細胞内の蛋白合成系に作用し、抗腫瘍効果を発揮する。このような効果を示すために、リポソーム製剤からの薬剤の放出とリポソーム製剤の血中滞留性との均衡を保つ必要がある。
本発明において、「放出」とは、リポソーム製剤に含まれる薬剤がリポソームから離脱することを意味する。
本発明のリポソーム製剤中に含まれる薬剤は、血漿中において、長時間高濃度で標的部位に暴露することで強い抗腫瘍活性を示すことから放出を制御することが重要となる。
【0033】
本発明のリポソーム製剤は、血漿中の薬剤濃度を高濃度で維持することができる。従来のリポソーム製剤は、血液中から速やかに消失するため、標的部位での暴露時間が短く、充分な効果を期待することは困難である。また、血液中からの速やかな消失は代謝器官である肝臓、脾臓などの臓器に対して薬剤を高濃度で暴露することになり、当該部位での副作用につながるため好ましくない。本発明のリポソーム製剤は、血漿中の薬剤濃度を高濃度で維持することができるため、肝臓、脾臓などの臓器に対する薬剤の暴露を軽減することができ、それに伴い、標的部位において薬剤を長時間暴露することができ、副作用を軽減することができることから好適である。
本発明において、投与後1時間における血漿中の薬剤濃度は、初期値の10%以上であり、好ましくは15%以上である。この「初期値」とは、本発明のリポソーム製剤を投与した直後の理論濃度であり、一般的に、宿主の体重から算出した全血漿量を用いて、投与液量で希釈されたと仮定して算出される。また、各時点の血漿中濃度は、初期値に対する比率として示すことができ、一般的に「% dose」として表記される。
本発明では、薬剤が所望の標的部位に長時間暴露するために使用される。したがって、本発明において、宿主の疾患の予防および/または治療のため、有効量の薬剤を担持するリポソーム製剤を宿主に投与することにより、宿主内で有効量の薬剤を放出し、標的部位に長時間高濃度で暴露するために、宿主(患者)に非経口的に全身あるいは局所的に投与することができる。投与対象の宿主としては、哺乳動物、好ましくはヒト、サル、ネズミ、家畜等が挙げられる。
【0034】
非経口的投与の経路としては、例えば点滴などの静脈注射(静注)、筋肉内注射、腹腔内注射、皮下注射を選択することができ、患者の年齢、症状により適宜投与方法を選択することができる。本発明の担体は、病気に既に悩まされる患者に、疾患の症状を治癒するか、あるいは少なくとも部分的に阻止するために十分な量で投与される。例えば、担体に封入される薬剤の有効投与量は、一日につき体重1kgあたり0.01mgから100mgの範囲で選ばれる。しかしながら、本発明の担体はこれらの投与量に制限されるものではない。投与時期は、疾患が生じてから投与してもよいし、あるいは疾患の発症が予測される時に発症時の症状緩和のために予防的に投与してもよい。また、投与期間は、患者の年齢、症状により適宜選択することができる。
具体的な投与方法としては、リポソーム製剤をシリンジや点滴によって投与することができる。また、カテーテルを患者または宿主の体内、例えば管腔内、例えば血管内に挿入して、その先端を標的部位付近に導き、当該カテーテルを通して、所望の標的部位またはその近傍あるいは標的部位への血流が期待される部位から投与することも可能である。
【実施例】
【0035】
次に実施例を挙げて本発明をさらに詳しく説明するが、本発明はこれら実施例に限定されるべきものではない。
各例で調製された薬剤封入リポソームの各濃度および粒子径は、以下のように求めた。
・ リン脂質濃度(mg/mL):高速液体クロマトグラフィーを用いて定量されるリポソーム懸濁液中でのリン脂質濃度。
・ コレステロール濃度(mg/mL):高速液体クロマトグラフィーを用いて定量されるリポソーム懸濁液中でのコレステロール濃度。
・ 総脂質濃度(mol/L):上記リン脂質濃度およびコレステロール濃度から算出される膜構成成分である脂質の合計モル濃度(mM)。この総脂質中には、PEGを導入するためのPEG誘導体中の脂質(例では、PEG−DSPE中のDSPE)は含まない。
・ 薬剤濃度(mg/mL):上記で得られた製剤をRO水(逆浸透膜浄水)でリン脂質濃度が約20mg/mLとなるように希釈した後、さらにメタノールで20倍希釈した溶液について、264nmでの吸光度を、紫外吸光光度計を用いて高速液体クロマトグラフィーにて定量した。内封されたKRN5500濃度を薬剤量(mg)/製剤全量(mL)で示す。
高速液体クロマトグラフィー試験条件:
カラム:内径6mm、長さ15cmのステンレス管に全多孔性球状シリカゲルを充填
(ナカライテスク(株)COSMOSIL 5C18−ARII)。
カラム温度:30℃付近
移動相:pH6.0のクエン酸緩衝液100mLに液体クロマトグラム用メタノール400mLを加え混合する。
流量:1.5mL/min
・ 薬剤担持量(薬剤/総脂質のモル比):リポソームに内封されたKRN5500濃度を上記総脂質濃度に対する上記薬剤濃度の比から、薬剤/総脂質のモル比で示す。
・ 粒子径(nm):リポソーム分散液20μLを生理食塩水3mLに希釈し、Zatasizer 3000HS(Malvern Instruments)またはZatasizer Nano ZS90で測定した平均粒子径。
以下に使用した各成分の略称および分子量を示す。
DSPC:(ジステアロイルフォスファチジルコリン、分子量790.2、日油)
HSPC:水素添加大豆フォスファチジルコリン(分子量790、リポイド(Lipoid)社製SPC3)
HEPC:水素卵黄大豆フォスファチジルコリン(分子量777、日油)
DPPC:(ジパルミトイルフォスファチジルコリン、分子量734.0、日油)
DMPC:(ジミリストイルフォスファチジルコリン分子量677.9、日油)
Chol:コレステロール(分子量388.66、Solvay)
PEG5000−DSPE:ポリエチレングリコール(分子量5000)−ジステアロイルフォスファチジルエタノールアミン(分子量6081、日油)
KRN5500(分子量589.7、協和発酵キリン(株))
【0036】
[実施例1]
(調製例1〜6)KRN5500リポソーム製剤の調製
KRN5500封入リポソーム製剤を達成しうる膜処方を確認するために、アシル鎖長の異なるリン脂質を用いてリポソーム化を試みた。また、異なるリン脂質/Chol比を有する膜処方についても試みた。調製例1、5、6は100mLスケールで調製し、調製例2、3,4は10mLスケールで製造した。なお、下記は100mLスケールでの調製法を示す。
(1)KRN5500リポソーム生成
各種類のリン脂質、コレステロール、KRN5500を表1に示す所定のモル比となるように秤量し、無水エタノール10mLを添加し、加温溶解した。
得られた脂質/薬剤混合エタノール溶液10mLに、約70℃に加温した内水相(pH6.5の10mMクエン酸一水和物/0.9%塩化ナトリウム溶液)90mLを添加し、超音波装置にて撹拌して粗リポソーム懸濁液を調製した。この粗リポソーム懸濁液を、約70℃に加温したエクストルーダー(The Extruder T.100、Lipexbiomembranes Inc.)に取り付けたフィルター(孔径0.2μm×3回、0.1μm×10回、Whatman社)を順次通し、リポソーム懸濁液を調製した。
(2)表面修飾
上記リポソーム懸濁液を加温状態で維持したまま、PEG5000−DSPEの水溶液(37.7mg/mL)を、表1に示すPEG導入率となるように直ちに添加し、加温撹拌することで、リポソームの膜表面(外表面)をPEG修飾した。加温終了後のリポソーム懸濁液は、速やかに氷冷した。
なお、上記PEG導入率(mol%)=(PEG5000−DSPE/総脂質)×100
である。ここでの総脂質は、PEG誘導体中の脂質(PEG5000−DSPE中のDSPE)は含まない。
(3)外液置換
上記氷冷したPEG修飾後のリポソーム懸濁液を、外水相溶液(10mMクエン酸一水和物/0.9%塩化ナトリウム溶液(pH6.5))を用いてクロスフローろ過システム(ビバフロー MW100,000)により外液置換を行った。なお、高濃度のKRN5500リポソーム製剤を得るために濃縮を行った(調製例1、5、6)。
(4)ろ過滅菌
外液置換後の上記リポソーム懸濁液を用いてろ過滅菌を行い無菌化し、最終製剤とした。ろ過滅菌後のリポソーム懸濁液を高速液体クロマトグラフィーを用いて、リン脂質、CholおよびKRN5500濃度を定量した。リン脂質濃度、Chol濃度の総和を総脂質濃度とし、封入されたKRN5500量を求めた。
上記で得られたKRN5500リポソーム製剤の膜組成比、薬剤担持量(薬剤濃度、薬剤/総脂質モル比)粒子径を表1に示す。
調製例1〜4に示すように、コレステロールを約半量含む膜処方の薬剤担持量は、リン脂質のアシル鎖長に関わらず、ほぼKRN5500/総脂質量=0.01(mol/mol)を示した。一方、コレステロールを含まない膜処方(調製例5および6)は、脂質/薬剤混合エタノール溶液の調製時において、仕込みのKRN5500量を約2倍に増やすことができるため、得られたリポソーム製剤の薬剤担持量も約2倍に増加した。
また、製剤化における脂質組成比の影響を検討した結果、表2に示すように、リン脂質のアシル鎖長および膜組成比によってリポソーム化が困難な膜処方があることが明らかとなった。リン脂質のアシル鎖長が長い場合は(ジステアロイル、パルミトイルステアロイル)コレステロールを含まなくてもリポソーム化することができるが、短い場合は(ジパルミトイル、ジミリストイル)、コレステロールを含まないとゲル化が生じ、製剤化できないことが明らかとなった。特にジミリストイル(C14)を用いる場合は、リン脂質に対して約半量のコレステロールが必要であることが明らかとなった。これは、コレステロールの膜安定化効果が影響していると考えられる。すなわち、アシル鎖長が長ければ疎水性相互作用が強まるため、コレステロールによる膜安定化効果がなくてもKRN5500を安定に内封することができると推測される。一方、アシル鎖長が短いと疎水性相互作用が弱まり、膜の流動性が大きくなるため、KRN5500を膜に安定に内封することができず、その結果、KRN5500自体の凝集が起こり、ゲル化してしまうと推測される。
【0037】
【表1】
【0038】
【表2】
【0039】
[実施例2]
本発明のリポソームにおいて、最大薬剤封入量を得るために必要な仕込み量を調べた。
(調製例7〜16)
HSPC/Chol(モル比)=54/46膜処方において、仕込みのKRN5500の量を、薬剤/総脂質(mol/mol)比で0.002、0.003、0.005、0.007、0.008、0.009、0.010、0.012、0.014、0.016、0.018、0.02となる量に変えた以外は、調製例1〜6と同様にしてKRN5500リポソーム製剤を得た。得られたKRN5500リポソーム製剤の薬剤/総脂質および粒子径を表3に示す。また、図1は薬剤担持量を、図2は封入効率のグラフを示す。
KRN5500仕込み量を薬剤/総脂質(mol/mol)=0.002から0.016に変えてリポソーム化の検討を行った結果、図1に示すように、仕込みのKRN5500量の増加と供に、封入量の増加が認められた。仕込みのKRN5500量をさらに増やすことが可能であれば、封入量もさらに増やせることが期待されるが、整粒化工程において目詰まりが発生したり、あるいはKRN5500が脂質エタノール溶液に溶解しなくなったりするため、これ以上仕込み量を増やすことは困難である。したがって、該処方における最大薬剤担持量は、総脂質に対して約1mol%であることが明らかとなった。この結果は他のリン脂質を用いても同様であった。また、KRN5500の封入効率は、仕込み量に関わらず、60〜80%の値を示した(図2)。
【0040】
【表3】
【0041】
[試験例1]薬物動態(血中滞留性)
調製例1、2、4、5、6で調製されたKRN5500リポソーム製剤およびKRN5500単体を、KRN5500量として0.2mg/kgでラットに投与した。投与後、0.25、1、2、4、6、8、12、24、48時間経過後に採血し、遠心分離(5,000 rpm、10分、4℃)して血漿を採取した。引き続き、血漿40μLにメタノール200μLを添加し、遠心分離(10,000rpm、10分、4℃)して上清を採取した。採取した溶液について、264nmでの吸光度を、紫外吸光光度計を用いて高速液体クロマトグラフィーにて定量し、各血漿中のKRN5500濃度を求めた。なお、高速液体クロマトグラフィーの試験条件は、リポソームに内封されたKRN5500濃度の条件と同様である。その結果を表4および図3に示す。
血漿中のKRN5500濃度は、KRN5500単体の場合には、投与後急激に減少し、0.25および1時間のみ検出された。一方、リポソーム製剤はいずれも、投与後48時間まで検出され、KRN5500単体に比べて顕著な滞留時間の延長が認められた。「% dose」で表記すると、KRN5500単体は投与1時間後、約5〜6%であるのに対して、リポソーム製剤は70〜80%であった。また、血漿中濃度−時間曲線下面積(AUC)は、リポソーム製剤はKRN5500単体に比べて60〜100倍大きかった。これらの結果より、本発明におけるリポソーム製剤は、いずれの膜処方においても、血漿中KRN5500濃度を長時間高濃度で維持することが可能であることを確認できた。ただし、コレステロールを含まないHSPC100mol%処方の血中滞留性は、コレステロールを含む処方と比べて、やや低下する傾向があった。特にPEG5000−DSPEの修飾率が0.75mol%の場合は、投与後、直ちに分布し、再び血中に現れる血中動態を示した。しかしながら、PEG5000−DSPEの修飾率を2.0mol%に増加することにより、初期に観察された分布が消失し、血中滞留性の改善が認められた。
【0042】
【表4】
【0043】
[試験例2]殺細胞効果
in vitroでの殺細胞効果評価用には、調製例1、2、3、6および下記に示す調製例17のリポソーム製剤を用いた。
(調製例17)
膜処方がDMPC/Chol(モル比)=54/46となるようにDMPC、CholおよびKRN5500を秤量し、調製例1〜6と同様にしてKRN5500リポソーム製剤を得た(100mLスケール)。得られたKRN5500リポソーム製剤の薬剤/総脂質および粒子径を表5に示す。
【0044】
【表5】
【0045】
図4は、調製例1、2、3、6、17にて調製されたKRN5500リポソーム製剤のin vitroでの殺細胞効果を示す。
ヒト大腸癌株Colo205を10%ウシ胎児血清を含むRPMI培地で、2000cells/90マイクロ L/wellとなるように96wellプレートに蒔き、37℃、5%CO2にて5時間培養した。
被験物質は、KRN5500はDMSOを、KRN5500リポソーム製剤はpH6.5の10mMクエン酸/0.9%塩化ナトリウム溶液を媒体とし、最終濃度(KRN5500として0から10000nMまで10濃度)の200倍の希釈系列を調製し、これをRPMI培地にて20倍希釈した被験物質溶液(検体)を調製した。
細胞を蒔いた96wellプレートに、検体を10マイクロ L/well添加し、37℃、5%CO2にて72時間培養した後、Cell Counting Kit(DOJINDO)を10マイクロ L/well添加し、再び37℃、5%CO2にて約20分間培養し、吸光度(450nm、参照波長650nm)を測定した。
細胞増殖抑制作用は、細胞(−)、培地+被験物質の媒体のみのwellの吸光度(A)を100%阻害、細胞(+)、培地+被験物質の媒体のみのwellの吸光度(B)を0%阻害とし、検体による細胞増殖抑制作用は、抑制作用(%)=100×(B−検体吸光度)/(B−A)で算出した。
膜組成がリン脂質/Chol比=54/46(モル比)のKRN5500リポソーム製剤の殺細胞効果は、リン脂質のアシル鎖長が短いほど活性が高かった(図4)。これは、リン脂質のアシル鎖長が短いほど、アシル鎖長とKRN5500の脂肪酸側鎖の疎水性相互作用が弱く、KRN5500がリポソームから放出しやすいためだと考えられる。また、コレステロールを含まないHSPC100mol%処方においても、KRN5500単体と同程度の高い活性が得られた。
【0046】
[試験例3]抗腫瘍効果(単回投与)
ヒト大腸癌細胞(CoL−1)における単回投与での抗腫瘍効果には、調製例1、5、17と下記に示す調製例18のリポソーム製剤を用いた。
(調製例18)
膜処方がHEPC/Chol(モル比)=54/46となるようにHEPC、CholおよびKRN5500を秤量し、調製例1〜6と同様にしてKRN5500リポソーム製剤を得た(100mLスケールで調製)。得られたKRN5500リポソーム製剤の薬剤/総脂質および粒子径を表6に示す。
【0047】
【表6】
【0048】
ヒト大腸癌株CoL−1腫瘍はヌードマウス(BALB/C、nu/nu、♀)の皮下に接種して通常飼育にて植え継いだ。試験用にヌードマウス背側部皮下に腫瘍断片を接種して通常飼育し、腫瘍体積が100mm3前後に達した段階で腫瘍体積が同程度となるようにn=4で群設定を行った(Day1)。
調製例1、5、17、18で調製したKRN5500リポソーム製剤およびKRN5500単体を、1mg/mLとなるように媒体〔KRN5500の媒体(N,N−ジメチルアセトアミド 3.75%、ポリソルベート80 3.0%、2−アミノエタノール 0.45%、生理食塩水92.8%)、リポソーム製剤の媒体(pH6.5の10mMクエン酸/0.9%塩化ナトリウム溶液)〕で調製し、10mg/kgで静脈内投与した。
KRN5500の媒体群およびリポソーム製剤の媒体群を対照群とした〔Control(Liposome)、Control(KRN5500単体)〕。
腫瘍体積は、Day1より2〜3日間隔で腫瘍の長径(Lmm)、短径(Wmm)、厚さ(Hmm)をノギスで測定し、L×W×H/2mm3で算出した。
図5は、投与後の経過日数における腫瘍体積を示す。KRN5500の媒体およびリポソーム製剤の媒体を投与したマウスを対照群とした[Control(Liposome),Control(KRN5500単体))。
ヒト大腸癌に対して、リポソーム製剤およびKRN5500単体は、いずれも対照群と比較して有意な腫瘍増殖抑制効果を示した。また、リポソーム製剤は、KRN5500単体に比べ高い持続的な抗腫瘍効果が得られた。特に、調製例6および17のリポソーム製剤の最大抗腫瘍効果はKRN5500単体と同程度であり、かつKRN5500単体よりも著しい持続効果が認められた。
【0049】
[試験例4]抗腫瘍効果(反復投与)
ヒト肺癌細胞株PC−9を10%ウシ胎児血清を含むRPMI培地にて培養し、PBSにて洗浄後、10%Tripsin−EDTA処理し、RPMI培地にて2回洗浄後、ヌードマウス(BALB/C、nu/nu、♀)の背側部皮下に5×106cells/100マイクロ L/個体となるように接種し通常飼育した。腫瘍体積が100mm3前後に達した段階で腫瘍体積が同程度となるようにn=4で群設定を行った(Day1)。
Day1より7日間隔で計4回(Day1、8、15、22)、調製例6ならびに17で調製したリポソーム製剤およびKRN5500単体をそれぞれの媒体で1mg/mLとなるように調製し、10mg/kgを静脈内投与した。KRN5500の媒体群およびリポソーム製剤の媒体群を対照群とした〔Control(Liposome)、Control(KRN5500単体)〕。
Day1より、2〜3日間隔で試験例3に準じて腫瘍体積を測定し、またマウス体重(g)を、体重測定値(g)−腫瘍体積(mm3)/1000で算出し、Day1の値を1とした体重変化率を指標とした。
投与後1、3、5、8、10、12、15、17、19、22、24日後に推定腫瘍体積およびマウスの体重を求めた。その結果を表7ならびに図6および図7に示す。
なお、相対腫瘍体積(RTV)および腫瘍増殖抑制率(TGIR)は下記の式を用いて算出した。
【0050】
相対腫瘍体積(RTV)=Day Xの腫瘍体積/Day 1の腫瘍体積
腫瘍増殖抑制率(TGIR、(%))=(1−被験物質投与群のRTV/Control群のRTV)×100
ヒト肺癌細胞に対して、リポソーム製剤およびKRN5500単体は、いずれも対照群と比較して有意な腫瘍増殖抑制効果を示した。さらに、リポソーム製剤はいずれの処方もKRN5500単体に比べて高い抗腫瘍効果が得られ、かつ持続的な効果が認められた。特にDMPC/Chol膜処方のリポソーム製剤は、最も高い抗腫瘍効果が得られた。また、リポソーム製剤はマウスの体重にほとんど影響を与えなかったのに対して、KRN5500単体は投与に伴う一過性の体重減少が観察された。これらの結果より、KRN5500をリポソーム化することで、抗腫瘍効果を高めることができ、かつ副作用も軽減できることが明らかとなった。尚、他の処方のリポソーム製剤もマウスの体重変化にはほとんど影響を及ぼさず、副作用を軽減できることが明らかとなった。また、ラット急性毒性試験の結果では、リポソーム化することによりKRN5500単体で認められた急性死を回避することができ、また肝臓の白色化を軽減できることが明らかとなった。
【0051】
【表7】
【特許請求の範囲】
【請求項1】
コレステロールが0〜50mol%および飽和脂肪酸のアシル鎖の鎖長がC14〜C18のいずれかであるリン脂質100〜50mol%を含有するリポソームに、化学式(I)で示されるスピカマイシン誘導体を有するリポソーム製剤:
【化1】
(式中、R1=H、R2=OHであって、Rは炭素数9〜15の直鎖のもしくは分岐したアルキルまたは炭素数10〜17の直鎖のアルケニルである。)。
【請求項2】
前記リポソーム製剤のリポソーム膜が、以下の1)〜3)のいずれかである請求項1に記載のリポソーム製剤:
1)リポソーム膜が、アシル鎖の鎖長が18である飽和脂肪酸を有するリン脂質を主構成成分として有する、
2)リポソーム膜が、アシル鎖の鎖長が16である飽和脂肪酸を有するリン脂質およびコレステロールを主構成成分として有し、両成分のモル比が80:20〜50:50である、
3)リポソーム膜が、アシル鎖の鎖長が14である飽和脂肪酸を有するリン脂質およびコレステロールを主構成成分として有し、両成分のモル比が60:40〜50:50である。
【請求項3】
前記リン脂質がフォスファチジルコリンである請求項1または2に記載のリポソーム製剤。
【請求項4】
前記、基R=CH3(CH2)8CH=CHCH=CH-であり、化学式(I)の化合物が(6−[4−de
oxy−4−(2E,4E)−tetradecadienoylglycyl]−amino−L−glycero−b−L−mannnoheptopyranosyl)amino−9H−purine)(KRN5500と称される)である請求項1〜3のいずれかに記載のリポソーム製剤。
【請求項5】
前記リポソーム製剤が親水性高分子鎖を有する請求項1〜4のいずれかに記載のリポソーム製剤。
【請求項6】
化学式(I)で示されるスピカマイシン誘導体の血漿中濃度−時間曲線下面積が、該スピカマイシン誘導体単体に比べて45倍以上増強することができる請求項1〜5のいずれかに記載のリポソーム製剤。
【請求項1】
コレステロールが0〜50mol%および飽和脂肪酸のアシル鎖の鎖長がC14〜C18のいずれかであるリン脂質100〜50mol%を含有するリポソームに、化学式(I)で示されるスピカマイシン誘導体を有するリポソーム製剤:
【化1】
(式中、R1=H、R2=OHであって、Rは炭素数9〜15の直鎖のもしくは分岐したアルキルまたは炭素数10〜17の直鎖のアルケニルである。)。
【請求項2】
前記リポソーム製剤のリポソーム膜が、以下の1)〜3)のいずれかである請求項1に記載のリポソーム製剤:
1)リポソーム膜が、アシル鎖の鎖長が18である飽和脂肪酸を有するリン脂質を主構成成分として有する、
2)リポソーム膜が、アシル鎖の鎖長が16である飽和脂肪酸を有するリン脂質およびコレステロールを主構成成分として有し、両成分のモル比が80:20〜50:50である、
3)リポソーム膜が、アシル鎖の鎖長が14である飽和脂肪酸を有するリン脂質およびコレステロールを主構成成分として有し、両成分のモル比が60:40〜50:50である。
【請求項3】
前記リン脂質がフォスファチジルコリンである請求項1または2に記載のリポソーム製剤。
【請求項4】
前記、基R=CH3(CH2)8CH=CHCH=CH-であり、化学式(I)の化合物が(6−[4−de
oxy−4−(2E,4E)−tetradecadienoylglycyl]−amino−L−glycero−b−L−mannnoheptopyranosyl)amino−9H−purine)(KRN5500と称される)である請求項1〜3のいずれかに記載のリポソーム製剤。
【請求項5】
前記リポソーム製剤が親水性高分子鎖を有する請求項1〜4のいずれかに記載のリポソーム製剤。
【請求項6】
化学式(I)で示されるスピカマイシン誘導体の血漿中濃度−時間曲線下面積が、該スピカマイシン誘導体単体に比べて45倍以上増強することができる請求項1〜5のいずれかに記載のリポソーム製剤。
【図1】

【図2】

【図3】
【図4】

【図5】

【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2012−236772(P2012−236772A)
【公開日】平成24年12月6日(2012.12.6)
【国際特許分類】
【出願番号】特願2009−223004(P2009−223004)
【出願日】平成21年9月28日(2009.9.28)
【出願人】(000109543)テルモ株式会社 (2,232)
【出願人】(000001029)協和発酵キリン株式会社 (276)
【Fターム(参考)】
【公開日】平成24年12月6日(2012.12.6)
【国際特許分類】
【出願日】平成21年9月28日(2009.9.28)
【出願人】(000109543)テルモ株式会社 (2,232)
【出願人】(000001029)協和発酵キリン株式会社 (276)
【Fターム(参考)】
[ Back to top ]