
テノホビルジソプロキシル−ヘミフマル酸共結晶
本発明は、共結晶TDFA 2:1と表す、テノホビルジソプロキシルフマレート(テノホビルDF)の新規な結晶形態、その調製方法、並びに医薬応用、特に抗HIV薬剤におけるその使用を提供する。結晶形態TDFA 2:1は、別の抗HIV薬剤、例えばエファビレンツ、エムトリシタビン、リトナビル、及び/またはTMC114と組み合わせて使用することができる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、テノホビルジソプロキシルとフマル酸との、モル比2:1での新規な共結晶組成物、その調製方法、及びその形成、並びに医薬品分野、特に抗ウィルス医薬品の分野における応用に関する。
【背景技術】
【0002】
テノホビルジソプロキシルフマレート(DF)は、米国において、単独でまたは別の抗レトロウィルス薬と組み合わせてHIV-I感染の治療用に認可された、ヌクレオチド系逆転写酵素阻害剤である。テノホビルジソプロキシルDFは、商品名VIREAD(登録商標)(Gilead Science, Inc.)として市販されており、別の抗ウィルス剤との組み合わせとしてTRUVADA(登録商標)及びATRIPLA(登録商標)抗HIV薬剤として市販されている。
【0003】
開発されている抗HIV薬剤の中には、HIV逆転写酵素(RT)またはプロテアーゼ酵素をターゲットとするものがあるが、これらの酵素はいずれもウィルスの複製に必要とされるものである。RT阻害剤の例には、ヌクレオシド/ヌクレオチドRT阻害剤(NRTI類)及び非ヌクレオチド系RT阻害剤(NNRTI類)が含まれる。現在、HIV感染患者は、3つの複合薬で規定通り治療されている。(少なくとも)3つのNRTI類を含む投薬計画、2つのNRTI類と1つまたは2つのプロテアーゼ阻害剤(PI)類との複合薬を含む投薬計画、あるいは2つのNRTI類と1つのNNRTIとの複合薬を含む投薬計画が、広く用いられている。これらの複合薬において2つ以上のPI類が使用される場合には、PI類の1つはしばしばリトナビルであり、これは治療量以下の低い用量で与えられ、投薬計画内の別のPI類の除去の効果的な阻害剤として作用して、ウィルスの最大限の抑制をもたらし、これにより耐性発現を低減する。
【0004】
臨床研究により、これらの抗HIV薬剤の3剤複合薬は、疾病進行及び死の回避に、1つの薬剤を単独でまたは2剤複合薬を用いるよりも格段に効果的であることが判明している。こうした薬剤の様々な複合薬を用いた、薬剤複合物の多数の研究により、こうした複合薬によってHIVに感染した人々において疾病進行及び死が大幅に低減されることが確立している。現在一般的に抗HIV薬剤の複合薬に与えられている名称は、HAART(高活性レトロウィルス療法)である。
【0005】
テノホビルDF、テノホビルジソプロキシル、TDF、ビス-POC-PMPAとしても既知である(米国特許第5,935,946号明細書、第5,922,695号明細書、第5,977,089号明細書、第6,043,230号明細書、第6,069,249号明細書)、テノホビルジソプロキシルフマレートは、テノホビルのプロドラッグ塩である。テノホビルジソプロキシルフマレートの化学名は、9-[(R)-2-[[ビス[[(イソプロポキシカルボニル)オキシ]メトキシ]ホスフィニル]-メトキシ]プロピル]アデニンフマレート(1:1)である。CAS登録番号は、202138-50-9である。これは、C19H30N5O10P・C4H4O4の分子式及び635.52の分子量を有する。これは以下の構造式:
【化1】

を有する。
【0006】
テノホビルDFの結晶形態は、とりわけ、国際特許公報第99/05150号、欧州特許第998480号明細書、及び米国特許第5935946号明細書に記載されている。この結晶形態(Gilead 1)は、約4.9、10.2、10.5、18.2、20.0、21.9、24.0、25.0、25.5、27.8、30.1、及び30.4のXRPDピークを有することにより特徴付けられる。更にまた、これらの結晶は、不透明または乳白色であって、開始温度が約116℃である、約118℃でのDSC吸収ピークを示し、且つそのIRスペクトルは、カイザーで表される、およそ3224、3107-3052、2986-2939、1759、1678、1620、1269、及び1102に特性帯を示すと記載されている。嵩密度は、約0.15-0.30g/mL、通常は約0.2-0.25g/mLと記載されている。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】米国特許第5,935,946号明細書
【特許文献2】米国特許第5,922,695号明細書
【特許文献3】米国特許第5,977,089号明細書
【特許文献4】米国特許第6,043,230号明細書
【特許文献5】米国特許第6,069,249号明細書
【特許文献6】国際特許公報第99/05150号
【特許文献7】欧州特許第998480号明細書
【特許文献8】米国特許第5935946号明細書
【発明の概要】
【発明が解決しようとする課題】
【0008】
テノホビルDFを含む幾つかの市販製品を分析したところ、これらが、固体形態のテノホビルDFの様々な割合の混合物を含むことが判明した。本発明者により、市販製品中の固体形態のテノホビルDFが、一般的に少なくとも2つの形態の混合物である印が発見されている。これらの形態の1つが、応力を受けた場合、例えば高温下及び/または高湿度下におかれた場合、その結晶形態から別の形態への転化を経ることもまた判明している。本発明者は、水の存在が1つの形態から別の形態への転化を誘発または促進すると考えている。このことは、市販製品中に現在使用されている固体形態が安定でないか、または少なくとも低い安定性を有することを示唆する。市販製品中におけるテノホビルジソプロキシルのフマル酸に対するモル容積比は、一般的に1:1とされる。
【課題を解決するための手段】
【0009】
本発明は、テノホビルジソプロキシルとフマル酸との、モル比2:1での新規な共結晶(TDFA 2:1)に関する。本発明は、1:1のフマレート塩である、テノホビルDFとは異なる。本発明のTDFA 2:1共結晶は、現在既知であるテノホビルDFの結晶形態(Gilead 1)よりも安定性が高く、且つ吸湿性が低い。
る。
【図面の簡単な説明】
【0010】
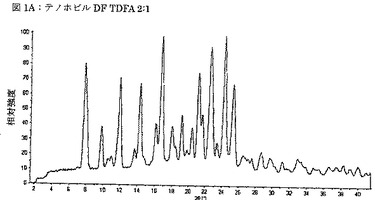
【図1A】図1Aは、テノホビルジソプロキシルとフマル酸との2:1モル比での共結晶(TDFA 2:1)のX線粉末回折パターンを示す図である。
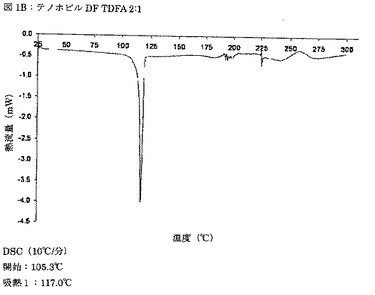
【図1B】図1Bは、テノホビルジソプロキシルとフマル酸との2:1モル比での共結晶(TDFA 2:1)のDSCサーモグラムを示す図である。
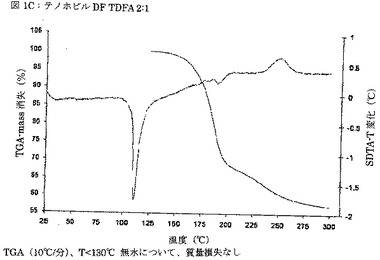
【図1C】図1Cは、テノホビルジソプロキシルとフマル酸との2:1モル比での共結晶(TDFA 2:1)のTGAサーモグラムを示す図である。
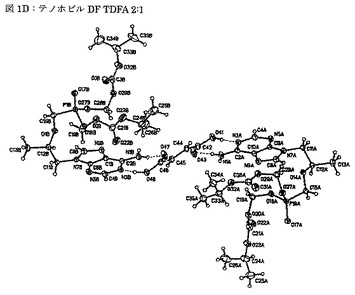
【図1D】図1Dは、単結晶のデータから測定される、テノホビルジソプロキシルとフマル酸との2:1モル比での共結晶(TDFA 2:1)由来の遊離塩基−遊離酸−遊離塩基体の分子構造を示す図である。
【図1E】図1Eは、テノホビルジソプロキシルとフマル酸との2:1モル比での共結晶(TDFA 2:1)の結晶充填を示す図である。
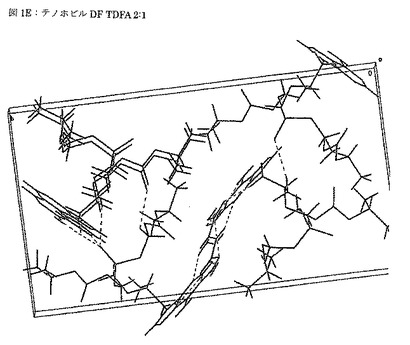
【図1F】図1Fは、テノホビルジソプロキシルとフマル酸との2:1モル比での共結晶(TDFA 2:1)のラマンスペクトルを示す図である。
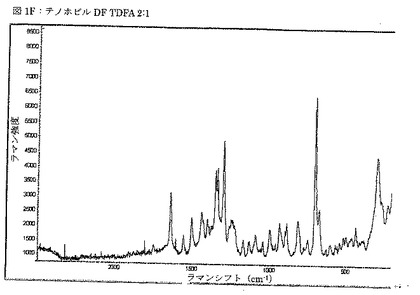
【図2A】図2Aは、粉砕したViread錠剤から得られるX線粉末回折パターンを示す図である。
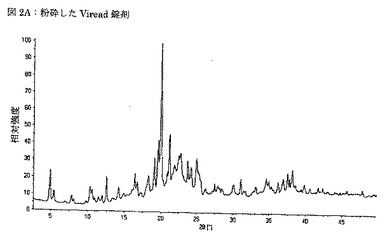
【図2B】図2Bは、コーティングを除去したVireadの錠剤から得られるX線粉末回折パターンを示す図である。
【図2C】図2Cは、粉砕したTruvada錠剤から得られたX線粉末回折パターンを示す図である。
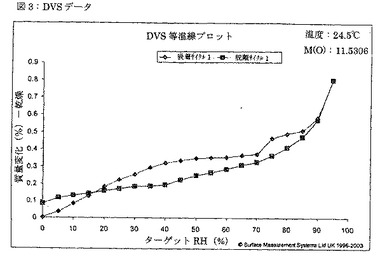
【図3】図3は、TDFA 2:1形態の吸着(ダイヤ形)及び脱離(四角形)挙動のDVSプロットを示す図である。
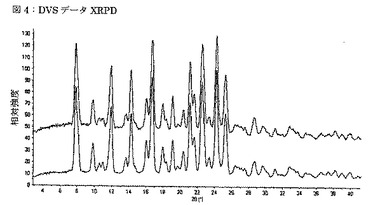
【図4】図4は、DVS測定前(上)及びDVS測定後(下)TDFA 2:1の実験的XRPDパターンを示す図である。
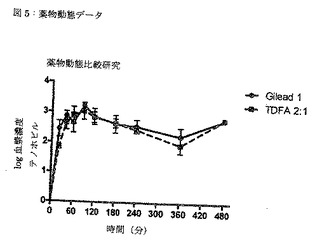
【図5】図5は、TDFAの生物学的同等性を示す、薬物動態データである。
【発明を実施するための形態】
【0011】
テノホビルジソプロキシル/フマル酸 共結晶(TDFA 2:1)
本発明は、2単位のテノホビルジソプロキシルが、1単位のフマル酸と、実験式C19H30N5O10P・C4H4O4をもって共結晶化した、テノホビルジソプロキシルとフマレートとの共結晶に関する。この共結晶は、テノホビルジソプロキシルのヘミフマル酸共結晶である。1つの態様では、本発明は、実質的に純粋な組成物、特にテノホビルジソプロキシルとフマル酸との、モル比2:1での共結晶(TDFA 2:1)を提供する。共結晶とは、1つまたは複数の分子性物質が、単位格子内に取り込まれた結晶体である。共結晶には、通常、塩類、例えばテノホビルDFが含まれず、こうした塩類は、逆帯電したイオン間及び溶媒和物間の静電結合をもたらすプロトン移動によって識別され、その結合機構は共結晶におけるものと同様であってもよいが、これらはその結晶化する際の溶媒と基質との会合である。例えば、Visheweshwar, P.; McMahon, J. A.; Bis, J. A.; Zaworotko, M. J. (2006) J. Pharm. Sci. 95(3), 499-516を参照のこと。
【0012】
上述の通り、本発明の新規な固体形態のTDFA 2:1は、独立して、実質的に純粋形態であり、好ましくは実質的に別のアモルファス及び/または結晶固体形態、例えば前述の従来技術に記載されている固体形態、すなわち本明細書中の別の箇所にも記載されるGilead 1またはULT-1を含まない。この点について、「実質的に含まない」とは、少なくとも約80%、85%、90%、95%、96%、97%、98%、または99%の純粋化合物に関する。この点について、「実質的に別のアモルファス及び/または結晶固体形態を含まない」とは、本発明による形態中に存在する、これらの別のアモルファス及び/または結晶固体形態が、約20%、15%、10%、5%、4%、3%、2%、1%以下であることを意味する。
【0013】
本発明の共結晶は、室温未満の温度、好ましくは120K前後の温度で共結晶である。本発明の共結晶は、より高い温度、例えば室温でも共結晶である。室温での単結晶構造及び実験的XRPDパターンにより、120Kと室温との間では構造相転移が全く起こっておらず、これらの温度でのXRPDパターンにおける相違は熱膨張によることが示される。以上に基づいて、TDFA 2:1は、室温(測定の地理的な位置によって15乃至40℃)では共結晶である。
【0014】
TDFA 2:1は、7.9、9.8、11.0、12.0、13.7、14.3、16.1、16.8、18.0、19.2、20.4、21.2、21.7、22.6、23.4、24.3、25.4、27.6度2θ±0.3度2θ、好ましくは±約0.2度2θ、更に好ましくは±約0.1度2θ、最も好ましくは±約0.05度2θからなる群より選択される、少なくとも1つ、好ましくは少なくとも2つ、より好ましくは少なくとも3つ、更にいっそう好ましくは少なくとも4つ、特に好ましくは少なくとも5つ、最も好ましくは6つのX線粉末回折ピークの選択によって特徴付けられる。好ましい実施態様では、少なくとも7つ、より好ましくは少なくとも8つ、更にいっそう好ましくは少なくとも9つ、特に好ましくは少なくとも10、最も好ましくは11のX線粉末回折ピークが上記群より選択される。より好ましい実施態様では、少なくとも12、より好ましくは少なくとも13、更にいっそう好ましくは少なくとも14、特に好ましくは少なくとも15、最も好ましくは16のX線粉末回折ピークが上記群より選択される。
【0015】
好ましくは、TDFAは、7.82、8.09、11.95、16.80、21.20、22.52、24.29度2θからなる群より選択される、少なくとも1つ、好ましくは少なくとも2つ、より好ましくは少なくとも3つ、更にいっそう好ましくは少なくとも4つ、特に好ましくは少なくとも5つ、最も好ましくは6つのX線粉末回折ピークの選択によって特徴付けられる。2θの位置は、室温で、1.54178Åの波長を用いて、TDFA 2:1の単結晶構造から算出される。実験的XRPDパターンにおいては、実験設定及びピーク重なりにより、以上に挙げた値から偏差があってもよい。
【0016】
TDFA 2:1は、下記のX線回折ピーク群によって、さらに任意に付随する強度によって、特徴付けることができる。
【表1】
【0017】
別の実施態様では、TDFA 2:1は、実質的に図1Aに従う、X線粉末回折パターンによって特徴付けられる。
別の実施態様では、TDFA 2:1は、実質的に図1Bに従う、DSCによって特徴付けられる。
別の実施態様では、TDFA 2:1形態は、実質的に図1Cに従う、TGAによって特徴付けられる。
別の実施態様では、本発明のTDFA 2:1形態は、開始温度が105.3℃である、117.0℃での特徴ピークを示すDSCによって特徴付けられるが実質的に図1Bに従う、DSCによって特徴付けられる。熱分析により、共結晶TDFA 2:1は非溶媒和であると結論づけられる。
【0018】
本発明は、一態様において、表Iに記載される適当な溶媒またはその混合物に、テノホビルDFを溶解させるかまたは混入し、溶媒の蒸発によりTDFA 2:1を結晶化させる工程を含む、共結晶TDFA 2:1の調製方法に関する。
【0019】
本発明は、別の態様において、表IIに記載される適当な溶媒またはその混合物に、テノホビルDFを溶解させるかまたは混入し、飽和溶液の冷却及び/または蒸発結晶化によりTDFA 2:1を結晶化させる工程を含む、共結晶TDFA 2:1の調製方法に関する。
【0020】
本発明は、一態様において、表IIIに記載される適当な溶媒またはその混合物に、テノホビルDFを溶解させるかまたは混入し、表IIIに記載される貧溶媒添加によりTDFA 2:1を結晶化させる工程を含む、共結晶TDFA 2:1の調製方法に関する。
【0021】
本発明は、別の態様において、本明細書中の別の箇所(溶媒に関する章)に概説される適当な溶媒またはその混合物にテノホビルDFを溶解させるかまたは混入し、スラリー結晶化及び/またはシード結晶化によりTDFA 2:1を結晶化させる工程を含む、共結晶TDFA 2:1の調製方法に関する。
【0022】
本発明の共結晶は、図1D及び/または1Eに図示され、且つ/または表2及び3に記載されるTDFA 2:1の単結晶構造に関する一態様によっても特徴付けられる。
【0023】
【表2】
【0024】
一態様では、本発明はさらに、(参照のために援用することとする、出願人による同時係属出願である米国第60/873,267号明細書に記載の通り、)実質的に純粋であり、好ましくはテノホビルDF形態のULT-1を含まないTDFA 2:1に関する。米国第60/873,267号明細書に記載のテノホビルDF形態のULT-1は、5.0、5.5、10.3、10.6、10.9、11.4、14.2、17.3、18.3、19.9、22.0、22.9、25.0、27.9、30.1度2θ±0.3度2θ、好ましくは±約0.2度2θ、更に好ましくは±約0.1度2θ、最も好ましくは±約0.05度2θからなる群より選択される、少なくとも1つ、好ましくは少なくとも2つ、より好ましくは少なくとも3つ、更にいっそう好ましくは少なくとも4つ、特に好ましくは少なくとも5つ、最も好ましくは6つのX線粉末回折ピークの選択によって特徴付けられる。好ましい実施態様では、少なくとも7つ、より好ましくは少なくとも8つ、更にいっそう好ましくは少なくとも9つ、特に好ましくは少なくとも10、最も好ましくは11のX線粉末回折ピークが上記群より選択される。より好ましい実施態様では、少なくとも12、より好ましくは少なくとも13、更にいっそう好ましくは少なくとも14、特に好ましくは少なくとも15のX線粉末回折ピークが上記群より選択される。
【0025】
本発明の好ましい実施態様では、TDFA 2:1は、固体形態であるテノホビルDF形態のULT-1を実質的に含まない。本発明の好ましい実施態様では、TDFA 2:1は、5.0及び/または5.5度2θ±0.1度2θにX線ピークを有することによって特徴付けられる固体形態を、実質的に含まない。別の好ましい実施態様では、TDFA 2:1は、4.9及び/または5.4度2θ±0.1度2θにX線ピークを有することによって特徴付けられる固体形態を、実質的に含まない。別の好ましい実施態様では、TDFA 2:1は、4.97及び/または5.44度2θ±0.1度2θにX線ピークを有することによって特徴付けられる固体形態を、実質的に含まない。
【0026】
一態様では、本発明は、実質的に純粋であり、好ましくは、(本明細書中の別の箇所及び出願人による同時係属出願である米国第60/873,267号明細書に記載の通り)テノホビルDF形態のULT-1から得られるTDFA 2:1形態を含む、医薬品組成物に関する。
【0027】
一態様では、本発明は、Cipla社より入手される出発物質テノホビルDFから、表I、II、及び/またはIIIの1つまたは複数に挙げられた有機溶媒、あるいはこれらの混合物からの再結晶により、2:1ヘミフマル酸共結晶を生成させる、TDFA 2:1形態の調製方法に関する。
【0028】
一態様では、本発明は、水性雰囲気における結晶化による、テノホビルDF形態からのTDFA 2:1の調製方法に関する。
【0029】
共結晶の単結晶が、水、メタノール、イソプロピルアセテート、(R)-(-)-2-オクタノール中のテノホビルDFの飽和溶液の、室温またはより低温、好ましくは5℃での緩慢蒸発によって得られた。別の実施態様では、飽和溶液を1℃/時乃至5℃/時の冷却速度で冷却した後、この温度に数日おく。表I、II、及びIIIに挙げた溶媒から、共結晶TDFA 2:1を得ることも可能である。
【0030】
溶媒
本発明のTDFA 2:1の調製方法の所定の実施態様では、蒸発結晶化、熱時濾過貧溶媒添加、シード結晶化及び/またはスラリー結晶化のための溶媒は、好ましくは、(R)-(-)-2-オクタノール、1,2-ジエトキシエタン、1,2-ジメトキシエタン、1,4-ジオキサン、1-ブタノール、1-ヘプタノール、1-ヘキサノール、1-メトキシ-2-プロパノール、1-ニトロプロパン、1-オクタノール、2,2,2-トリフルオロエタノール、2-ブタノン、2-エトキシエタノール、2-エトキシエチルアセテート、2-ヘキサノール、2-メトキシエタノール、2-ニトロプロパン、2-ペンタノール、2-プロパノール、4-ヒドロキシ-4-メチル-2-ペンタノン、アセトン、アセトニトリル、ブチロニトリル、シクロヘキサノール、シクロペンタノール、シクロペンタノン、ジエチレングリコールジメチルエーテル、ジメチルカーボネート、ジメチルカーボネート、エタノール、エチルホルメート、エチルアセテート、エチレングリコールモノブチルエーテル、ジクロロメタン、フルフリルアルコール、イソブタノール、イソプロピルアセテート、メタノール、メトキシエチルアセテート、メチルアセテート、メチルブチレート、メチルプロピオネート、2-メチル-4-ペンタノール、N,N-ジメチルアセトアミド、N,N-ジメチルホルムアミド、ニトロベンゼン、ニトロエタン、ニトロメタン、N-メチルピロリドン、プロピオニトリル、プロピルアセテート、プロピレングリコールメチルエーテルアセテート、tert-ブタノール、テトラヒドロフラン、テトラヒドロフルフリルアルコール、テトラヒドロピラン、水、及びこれらの混合物から成る群より選択される。
【0031】
本発明のTDFA 2:1の調製方法の所定の実施態様では、蒸発結晶化、熱時濾過貧溶媒添加、シード結晶化及び/またはスラリー結晶化のための溶媒は、更に好ましくは、(R)-(-)-2-オクタノール、1,2-ジエトキシエタン、1,2-ジメトキシエタン、1,4-ジオキサン、1-ブタノール、1-ニトロプロパン、1-プロパノール、2-ブタノン、2-エトキシエチルアセテート、2-メチル-4-ペンタノール、2-ニトロプロパン、2-プロパノール、アセトン、アセトニトリル、シクロペンタノール、エタノール、イソブタノール、イソプロピルアセテート、メタノール、メトキシ-2-1-プロパノール、メチルプロピオネート、N,N-ジメチルアセトアミド、N,N-ジメチルホルムアミド、ニトロメタン、tert-ブタノール、テトラヒドロフラン、水、1,2-ジクロロエタン、2,6-ジメチル-4-ヘプタノン、アミルエーテル、ブチルベンゼン、クロロホルム、ジクロロメタン、ヘキサフルオロベンゼン、n-ヘプタン、N-メチルピロリドン、tert-ブチルメチルエーテル、トルエン、シクロペンタノンから成る群より選択される。
【0032】
本発明のTDFA 2:1の調製方法の所定の実施態様では、熱時濾過結晶化のための溶媒は、好ましくは、(R)-(-)-2-オクタノール、1,2-ジエトキシエタン、1,2-ジメトキシエタン、1,4-ジオキサン、1-ブタノール、1-ニトロプロパン、1-プロパノール、2-ブタノン、2-エトキシエチルアセテート、2-メチル-4-ペンタノール、2-ニトロプロパン、2-プロパノール、アセトン、アセトニトリル、シクロペンタノール、エタノール、イソブタノール、イソプロピルアセテート、メタノール、メトキシ-2-1-プロパノール、メチルプロピオネート、N,N-ジメチルアセトアミド、N,N-ジメチルホルムアミド、ニトロメタン、tert-ブタノール、テトラヒドロフラン、水、及びこれらの混合物から成る群より選択される。
【0033】
本発明のTDFA 2:1の調製方法の所定の実施態様では、溶媒/貧溶媒結晶化のための溶媒は、好ましくは、1,2-ジクロロエタン、1,2-ジメトキシエタン、1,4-ジオキサン、2,6-ジメチル-4-ヘプタノン、2-ブタノン、アセトン、アセトニトリル、アミルエーテル、ブチルベンゼン、クロロホルム、シクロヘキサン、シクロヘキサン、ジクロロメタン、ヘキサフルオロベンゼン、メタノール、n-ヘプタン、ニトロメタン、N-メチルピロリドン、tert-ブチルメチルエーテル、テトラヒドロフラン、トルエン、水、及びこれらの混合物から成る群より選択される。
【0034】
本発明のTDFA 2:1の調製方法の所定の実施態様では、貧溶媒結晶化のための貧溶媒は、好ましくは、1,2-ジクロロエタン、2,6-ジメチル-4-ヘプタノン、アセトン、アミルエーテル、ブチルベンゼン、クロロホルム、シクロヘキサン、ジクロロメタン、ヘキサフルオロベンゼン、n-ヘプタン、ニトロメタン、tert-ブチルメチルエーテル、トルエン、及びこれらの混合物から成る群より選択される。
【0035】
本発明のTDFA 2:1の調製方法の所定の実施態様では、シード結晶化のための溶媒は、好ましくは、メタノール、水、1,4-ジオキサン、アセトニトリル、2-エトキシエチルアセテート、2-メチル-4-ペンタノール、テトラヒドロフラン、ブチルベンゼン、アミルエーテル、tert-ブチルメチルエーテル、シクロペンタノン、及びこれらの混合物から成る群より選択される。
【0036】
本発明のTDFA 2:1の調製方法の所定の実施態様では、スラリー結晶化のための溶媒は、好ましくは、水、メタノール、アセトニトリル、1,4-ジオキサン、及びこれらの混合物から成る群より選択される。
【0037】
医薬品製剤
本発明は、更に、テノホビルDFの新規な結晶形態を含む医薬品製剤に関する。
本発明の医薬品製剤は、本明細書中に開示されるTDFA 2:1を含む。本発明はまた、本発明による結晶形態を一つまたは複数含む医薬品組成物を提供する。本発明の製剤処方は、本発明による結晶形態の一つまたは複数を活性成分として、任意に一つまたは複数の別の結晶形態との混合物として含む。
【0038】
本発明による製剤処方は、TDFA 2:1形態に加えて、付加的な医薬品活性成分、好ましくは、抗HIV剤、更に好ましくは、エファビレンツ、エムトリシタビン、リトナビル、及び/またはTMC114をさらに含んでよい。
【0039】
活性成分に加えて、本発明の医薬品製剤は、一つまたは複数の賦形剤を含んでよい。賦形剤は、様々な目的で製剤に加えられる。
【0040】
希釈剤は、固形医薬品組成物の体積を増大させ、患者及び介護人にとってのこの組成物を含む医薬品投薬形態の取り扱いを、より容易にしうる。固形組成物の希釈剤には、例えば、マイクロクリスタリンセルロース(例えばAvicel(登録商標))、マイクロファインセルロース、ラクトース、澱粉、アルファ化澱粉、炭酸カルシウム、硫酸カルシウム、糖、デキストレート類、デキストリン、デキストロース、リン酸水素カルシウム二水和物、リン酸三カルシウム、カオリン、炭酸マグネシウム、酸化マグネシウム、マルトデキストリン、マンニトール、ポリメタクリレート類(例えばEudragit(登録商標))、塩化カリウム、粉末セルロース、塩化ナトリウム、ソルビトール、及びタルクが含まれる。
【0041】
投薬形態、例えば錠剤にコンパクト化された固形医薬品組成物は、その機能の中に、圧縮後に活性成分と別の賦形剤との互いとの結合を補助することを含む、賦形剤を含んでよい。固形医薬品組成物のための結合剤には、アカシア、アルギン酸、カーボマー(例えば、Carbopol)、カルボキシメチルセルロースナトリウム、デキストリン、エチルセルロース、ゼラチン、グアーガム、水添植物油、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース(例えば、Klucel(登録商標))、ヒドロキシプロピルメチルセルロース(例えば、Methocel(登録商標))、液体グルコース、ケイ酸アルミニウム・マグネシウム、マルトデキストリン、メチルセルロース、ポリメタクリレート類、ポビドン(例えば、Kollidon(登録商標)、Plasdone(登録商標))、アルファ化澱粉、アルギン酸ナトリウム、及び澱粉が含まれる。
【0042】
患者の胃の中のコンパクト化固形医薬品組成物の溶解速度は、この組成物への崩壊剤の添加によって増大されうる。崩壊剤には、アルギン酸、カルボキシメチルセルロースカルシウム、カルボキシメチルセルロースナトリウム(例えば、Ac-Di-Sol(登録商標)、Primellose(登録商標))、コロイド状二酸化ケイ素、クロスカルメロースナトリウム、クロスポビドン(例えば、Kollidon(登録商標)、Polyplasdone(登録商標))、グアーガム、ケイ酸アルミニウム・マグネシウム、メチルセルロース、マイクロクリスタリンセルロース、ポラクリリンカリウム、粉末セルロース、アルファ化澱粉、アルギン酸ナトリウム、澱粉グリコール酸ナトリウム(例えば、Explotab(登録商標))、及び澱粉が含まれる。
【0043】
流動促進剤は、非コンパクト化固形組成物の流動性を改善し、且つ投薬の正確性を改善するために加えることができる。流動促進剤として機能しうる賦形剤には、コロイド状二酸化ケイ素、三ケイ酸マグネシウム、粉末セルロース、澱粉、タルク、及びリン酸三カルシウムが含まれる。
【0044】
投薬形態、例えば錠剤が粉末組成物のコンパクト化によって製造される場合、この組成物には、パンチ及びダイから圧力がかけられる。賦形剤及び活性成分の中には、パンチ及びダイの表面に付着する傾向を有するものがあり、このため製品に孔が生じ、また別の表面凹凸が生じうる。滑剤を前記組成物に加えて、粘着性を低減し、製品のダイからの除去を容易にすることもできる。滑剤には、マグネシウムステアレート、カルシウムステアレート、グリセリルモノステアレート、グリセリルパルミトステアレート、水添蓖麻子油、水添植物油、鉱物油、ポリエチレングリコール、ナトリウムベンゾエート、ナトリウムラウリルスルフェート、ナトリウムステアリルフマレート、ステアリン酸、タルク、及び亜鉛ステアレートが含まれる。香味料及び調味料は、投薬形態を、患者にとってより口当たりの良いものにする。本発明の組成物に導入してもよい、医薬品向けの通常の香味料及び調味料には、マルトール、バニリン、エチルバニリン、メントール、クエン酸、フマル酸、エチルマルトール、及び酒石酸が含まれる。固形及び液状の組成物はまた、その外観を改善し、且つ/または患者による製品及び単位投与量レベルの確認を容易にするために、医薬品として許容されるいかなる着色剤を使用して染色してもよい。
【0045】
本発明の液状の医薬品組成物中では、本発明による結晶形態及び別のあらゆる固形賦形剤が、液状担体、例えば、水、植物油、アルコール、ポリエチレングリコール、プロピレングリコール、グリセリン、またはこれらの混合物中に、本明細書中に記載の結晶形態が維持される、すなわち溶解しない限りは、懸濁している。
【0046】
液状医薬品組成物は、乳化剤を含んで、液状担体に溶解しない活性成分または別の賦形剤を、この組成物全体に均一に分散させてよい。本発明の液状組成物中に使用してよい乳化剤には、例えば、ゼラチン、卵黄、カゼイン、コレステロール、アカシア、トラガカンス、ツノマタ、ペクチン、メチルセルロース、カーボマー、セトステアリルアルコール、及びセチルアルコールが含まれる。
【0047】
本発明の液状医薬品組成物は、増粘剤をさらに含んで、製品の口当たりを改善するか、且つ/または胃腸管の内壁を覆ってよい。こうした作用剤には、アカシア、アルギン酸ベントナイト、カーボマー、カルボキシメチルセルロースカルシウムもしくはナトリウム、セトステアリルアルコール、メチルセルロース、エチルセルロース、ゼラチングアーガム、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、マルトデキストリン、ポリビニルアルコール、ポビドン、プロピレンカーボネート、プロピレングリコールアルギネート、ナトリウムアルギネート、ナトリウム澱粉グリコレート、澱粉トラガカンス、及びキサンタンガムが含まれる。
【0048】
甘味料、例えば、ソルビトール、サッカリン、ナトリウムサッカリン、スクロース、アスパルテーム、フルクトース、マンニトール、及び転化糖を加えて、味を改善してよい。保存料及びキレート剤、例えば、アルコール、ナトリウムベンゾエート、ブチル化ヒドロキシルトルエン、ブチル化ヒドロキシアニソール、及びエチレンジアミンテトラ酢酸を、摂取に安全なレベルで加えて、貯蔵安定性を改善してもよい。本発明によれば、液状組成物は、グルコン酸、乳酸、クエン酸、または酢酸、ナトリウムグルコネート、ナトリウムラクテート、ナトリウムシトレート、またはナトリウムアセテートをさらに含んでよい。使用される賦形剤及びその量の選択は、経験及び標準的操作を勘案し、且つ当該分野の参考文献に基づいて、製剤研究者によって容易に決定されうる。
【0049】
眼または別の外部組織、例えば、口及び皮膚の感染のためには、この製剤は、好ましくは、活性成分を、例えば、0.01乃至10質量%の量で含む(0.1質量%刻みで0.1乃至5質量%の範囲で、例えば0.6質量%、0.7質量%の活性成分を含む)、好ましくは0.2乃至3質量%、最も好ましくは2質量%の量で含む、局所用軟膏またはクリームとして適用される。軟膏として製剤化される場合には、活性成分を、パラフィン系または水混和性のいずれの軟膏ベースと共に用いてもよい。
【0050】
あるいはまた、活性成分を、水中油型クリームベースを用いてクリーム中に製剤化してよい。
【0051】
所望により、クリームベースの水性相は、例えば、少なくとも30質量%の多価アルコール、すなわち、二つ以上のヒドロキシル基を有するアルコール、例えば、プロピレングリコール、ブタン1,3-ジオール、マンニトール、ソルビトール、グリセロール、及びポリエチレングリコール(PEG 400を含む)、及びこれらの混合物を含んでよい。局所用製剤は、望ましくは、皮膚または別の罹患した領域からの、活性成分の吸収または浸透を促進する化合物を含んでよい。こうした皮膚透過促進剤には、ジメチルスルホキシド及び関連類縁体が含まれる。
【0052】
本発明のエマルションの油性相は、既知の成分から既知の方法で構成されてよい。この相は一つの乳化剤(エマルジェントとしても既知)のみを含んでよい一方で、望ましくは、少なくとも一つの乳化剤の、脂肪または油、あるいは脂肪及び油の両方との混合物を含む。好ましくは、親水性乳化剤が、安定化剤として作用する親油性乳化剤と共に導入される。油と脂肪とをいずれも導入することもまた好ましい。安定化剤と共にまたは安定化剤なしに、乳化剤は乳化ワックスを構成し、ワックスと油及び脂肪とは、共に乳化軟膏ベースを構成するが、これはクリーム製剤の油性分散相を形成する。
【0053】
本発明の製剤中における使用に適当なエマルジェント及びエマルション安定化剤には、Tween8 60、Spans 80、セトステアリルアルコール、ベンジルアルコール、ミリスチルアルコール、グリセリルモノステアレート、及びナトリウムラウリルスルフェートが含まれる。
【0054】
前記製剤に適当な油または脂肪の選択は、所望の化粧品特性の達成に基づく。然るに、クリームは、好ましくは、チューブまたは別の容器からの漏出をさけるために適当なコンシステンシーを有する、非脂肪性、非着色性、及び洗浄可能な製品であるべきである。
【0055】
直鎖状または分枝状である、一塩基性もしくは二塩基性のアルキルエステル類、例えば、ジイソアジペート、イソセチルステアレート、ココナッツ脂肪酸類のプロピレングリコールジエステル、イソプロピルミリステート、デシルオレエート、イソプロピルパルミテート、ブチルステアレート、2-エチルヘキシルパルミテート、または、Crodamol CAPとして既知の分枝鎖エステル類のブレンドを使用してよく、最後の3つが好ましいエステル類である。これらは、要求される特性に応じて、単独でまたは組合せとして使用してよい。あるいはまた、高融点脂質、例えば白色軟質パラフィン及び/または流動パラフィン、あるいは別の鉱物油が使用可能である。
【0056】
眼への局所適用に適当な製剤には、活性成分が適当な担体中、特に、前記活性成分のための水性溶媒中に、溶解または懸濁された、点眼薬がさらに含まれる。活性成分は、適切には、こうした製剤中に、0.01乃至20質量%の濃度で存在し、0.1乃至10質量%である態様もあれば、約1.0質量%である態様もある。
【0057】
口への局所適用に適当な製剤には、調味ベース中、通常は、スクロース及びアカシアまたはトラガカンス中に、活性成分を含むロゼンジ類;不活性ベース中、例えば、ゼラチンとグリセリン、またはスクロースとアカシア中に、活性成分を含むトローチ類;及び適当な液状担体中に活性成分を含むマウスウォッシュ類が含まれる。
【0058】
直腸投与のための製剤は、例えば、ココアバターまたはサリチレートを含む適当なベースと共に、坐薬として提示してよい。
【0059】
担体が固形である、経鼻または吸入投与に適当な製剤には、例えば、1乃至500ミクロンの範囲の粒径(5ミクロン毎に20乃至500ミクロンの範囲、例えば30ミクロン、35ミクロン等の粒径を含む)を有する粉末が含まれる。担体が液状であって、経鼻スプレーまたは点鼻薬に適当な製剤には、活性成分の水性または油性の溶液が含まれる。
【0060】
エアロゾル投与に適当な製剤は、定法により調製してよく、別の治療剤と共に送達されてよい。吸入療法剤は、定量吸入器によって容易に投与される。
【0061】
膣内投与に適当な製剤は、活性成分に加えて、当業者に適当であることが知られた担体を含む、ペッサリー類、タンポン類、クリーム類、ゲル類、ペースト類、フォーム類、またはスプレー製剤として提示してよい。
【0062】
本発明の固形組成物には、粉末、粒子、凝集体、及びコンパクト化組成物が含まれる。用量には、経口、口腔、直腸、非経口(皮下、筋内、及び静脈内を含む)、吸入、眼科投与に適当な用量が含まれる。所与のあらゆる場合において、最適な投与は、治療しようとする状態の性質及び重篤度に依存するが、本集め胃の最も好ましい経路は経口である。用量は、単位用量形態で従来提示されているものでよく、医薬品分野において周知のいかなる方法で調製してもよい。
【0063】
投与形態には、錠剤、粉末、カプセル、坐薬、サシェ、トローチ、及びロゼンジ、ならびに液状シロップ、懸濁液、及びエリキシル剤が含まれる。
【0064】
本発明の投与形態には、前記組成物を、好ましくは粉末化もしくは粒状化した本発明の固形組成物を、硬質または軟質シェル内に含むカプセルであってよい。このシェルは、ゼラチン製であってよく、任意に、グリセリン及びソルビトール等の可塑剤及び乳白剤または着色剤を含む。
【0065】
活性成分及び賦形剤は、当業者に既知の方法によって、組成物及び投薬形態に製剤化してよい。錠剤化またはカプセル充填のための組成物は、湿式造粒法によって調製してよい。湿式造粒法では、粉末形態の活性成分及び賦形剤のいくつかまたは全てをブレンドし、その後、当該粉末を粒子に凝集させる液体、典型的には水の存在下で混合する。粒子を、篩にかけ、且つ/または粉砕して乾燥させ、その後、篩にかけ、且つ/または粉砕して所望の粒径とする。その後粒子を錠剤化/圧縮するか、あるいは錠剤化の前に別の賦形剤、例えば、流動促進剤及び/または滑剤を、加えてよい。
【0066】
錠剤化組成物は、乾式混合によって定法で調製してよい。例えば、活性剤と賦形剤とのブレンド組成物を、スラグまたはシート状にコンパクト化した後、コンパクト化粒子状に粉砕してもよい。コンパクト化粒子は、次いで錠剤状に圧縮してよい。
【0067】
乾式造粒に代えて、ブレンド組成物を、直接圧縮技術を用いて、直接コンパクト化投薬形態に圧縮してもよい。直接圧縮によれば、粒子のない、より均一な錠剤が製造される。直接圧縮錠剤化に特に適した賦形剤には、マイクロクリスタリンセルロース、スプレー乾燥ラクトース、リン酸水素カルシウム二水和物、及びコロイド状シリカが含まれる。これら及び別の賦形剤の、直接圧縮錠剤化における適切な使用は、特に直接圧縮錠剤化の製剤研究の経験及び技術を有する当業者には既知である。
【0068】
本発明のカプセル充填物は、錠剤化に関して記載された前述のあらゆるブレンド及び粒子を含んでよいが、これらには最後の錠剤化工程は行わない。
【0069】
さらにまた、本発明による結晶形態は、哺乳類、好ましくはヒトへの注入による投与のために製剤化することができる。本発明による結晶形態は、例えば、注入のための粘性の液状溶液または懸濁液として製剤化してよい。製剤は、溶媒を含んでよい。こうした溶媒のための検討事項の中には、様々なpHレベルでの該溶媒の物理的及び化学的安定性、粘度(注射針通過性をもたらす)、流動性、沸点、混和性、及び純度が含まれる。適当な溶媒には、アルコールUSP、ベンジルアルコールNF、ベンジルベンゾエートUSP、及び蓖麻子油USPが含まれる。この製剤には、付加的物質、例えば、バッファー、溶解剤、酸化防止剤等をさらに加えてよい。Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery Systems, 7th Ed.
【0070】
本発明はまた、本発明による結晶形態を必要とする哺乳類、好ましくはヒトの治療方法に使用しようとする前記結晶形態を、任意に別の多形相または共結晶と共に含む医薬品製剤を提供する。本発明の医薬品組成物は、結晶形態のTDFA 2:1を含む。本発明による結晶形態は、こうした医薬品組成物の治療のための有効量を、本明細書中に前述される病気に罹患した哺乳類に投与する工程を含む、哺乳類の治療方法において使用してよい。本発明は、さらに、本明細書中に前述される病気、特にHIVの治療のための薬剤の調製のための、本発明の結晶形態の使用に関する。
【0071】
所定の好ましい実施態様に関して本発明を説明したが、別の実施態様は、当業者が本明細書を勘案することによって明らかになるであろう。本発明は、本発明の化合物の調製を詳細に説明する、以下の実施例を参照して更に規定される。当業者には、原材料と方法とのいずれについても、本発明の範囲から逸脱することなく多数の変形を実施しうることが明らかであろう。
【実施例】
【0072】
(実験条件)
X線粉末回折:
XRPDパターンが、Avantium technologies社(オランダ)製のT2ハイスループットXRPD装置を使用して得られた。Hi-Starエリア型検出器を装備した、Bruker GADDS回折計に、プレートを取り付けた。XRPDプラットフォームに、長い格子面間隔についてはベヘン酸銀を用い、短い格子面間隔についてはコランダムを用いて較正した。データ収集を、1.5°乃至41.5°の2θ領域内で、単色CuK(α)線(1.54178Å)を用いて室温で行った。各ウェルの回折パターンを、二つの2θ範囲(第一フレームについては1.5°≦2θ≦21.5°、第二フレームについては19.5°≦2θ≦41.5°)において、各フレームについて120秒の露光時間で収集する。当業者であれば、器具、試料調製、または別の要因における差異により、実験差異が生じうることを理解する。典型的には、XRPDデータは、約0.3度2θ、好ましくは約0.2度、更に好ましくは0.1度、更に一層好ましくは0.05度の変動で収集される。これは、X線ピークが重なっていると考えられる場合に影響を与える。
【0073】
高分解能X線粉末回折:
高分解能粉末パターンを、LynxEye固体検出器を装備した、Brag-Brentano配置のD8 Advanceシステムで収集した。データ収集のために使用された光は、ゲルマニウム結晶によって単色化されたCuK(α1=1.54056Å)であった。パターンを、約2-4°2θから開始して約60-65°2θまでの様々な2θ範囲で、0.04-0.16°2θの範囲のステップで、更なる処理を行わずに収集した。全てのパターンを室温、およそ295Kでとった。
【0074】
単結晶X線回折:
適当な単結晶を選択してガラスファイバーに接着し、これをX線回折ゴニオメーターに載せた。X線回折データを、三つの結晶について、120Kの温度及び室温で、KappaCCDシステム及びFR590 X線発生装置(Bruker Nonius社(オランダ、デルフト)製)により発生させたMoKα光を用いて収集した。単位セルパラメータ及び結晶構造を、ソフトウェアパッケージMaXusを使用して決定し、最適化した。
【0075】
熱分析:
融解特性を、熱流束DSC822e機器(Mettler-Toledo GmbH(スイス)社製)で記録した、DSCサーモグラムから得た。DSC822eを、インジウムの小片(融点=156.6℃;デルタ-H(f)=28.45J/g)を用いて温度及びエンタルピーについて較正した。試料を、標準的な40マイクロリットルのアルミニウムパンに封入し、DSCにおいて25℃乃至300℃に、20℃/分の加熱速度で加熱した。流速50ml/分で無水N2気体を用いて、測定の間、DSC装置をパージした。
【0076】
結晶からの水損失または溶媒による質量損失を、TGA/SDTAによって決定した。試料質量を、TGA/SDTA851e機器(Mettler-Toledo GmbH(スイス)製)で加熱する間に観察することにより、質量対温度の曲線が得られた。TGA/SDTA851eを、インジウム及びアルミニウムを用いて温度について較正した。試料を100マイクロリットルのアルミニウムるつぼに計量して仕込み、密閉した。封にピンで孔を空け、このるつぼを、TGAにおいて、20℃/分の加熱速度で25℃乃至300℃に加熱した。無水N2気体を用いてパージした。DSCに基づく融点決定は、±2.0℃、好ましくは1.0℃の変動性を有する。
【0077】
ラマン分光法:
ラマンスペクトルを、ラマン顕微鏡mW(Kaiser Opticals Inc製)で、0.96cm-1の分解にて、780nmのレーザー及び100の出力を用いて収集した。
【0078】
(実施例)
結晶化実験のための出発物質は、Cipla Ltd.(インド、ムンバイ)製の研究試料として得た。
高分解能X線回折計を使用する、幾つかの市販試料の分析:
テノホビルの市販試料を、地域の薬局(Viread and Truvada)から入手し、錠剤の表面から擦過及び研磨によってコーティングを注意深く除去して、コーティング物質がX線回折パターンに影響しないようにした。Vireadについての二つのXRPDパターンを、別々に調製した試料から、高分解能X線回折計を用いて収集した。第一の試料は、穏やかに粉砕した錠剤から調製し、第二の試料は、コーティング除去及び表面平坦化の後に粉砕されない錠剤から調製した。いずれの試料のXRPDパターンも、第一の試料の粉砕によって誘発される、構造相転移が全く起こっていないことを示した。
【0079】
VireadのX線分析により、これがテノホビルDFをGilead形態1(米国特許第5,935,946号に記載)及びテノホビルジソプロキシルフマレートの共結晶であるTDFA 2:1を含むことが示された。上述の全てのXRPDパターンは、いずれの錠剤においても賦形剤として使用された、ラクトース一水和物の存在を更に示した。表3Aには、粉砕したViread錠剤のXRPDパターンの2θ位置が第一列に、更にGilead形態1(米国特許第5,935,946号)のピーク位置が第二列に、使用された出発物質もしくは実験のピーク位置が第三列に、室温での単結晶構造に基づいて算出されたTDFA 2:1のピーク位置が第四列に、更に、Cambridge Structure Database (REFCODE LACTOS01)に見られる単結晶構造に基づいて算出されたラクトース一水和物のピーク位置が第五列にまとめられている。
【0080】
その粉砕錠剤のXRPDパターンを1つ収集した、Truvada(詳細な表は記載せず)のXRPDパターンを研究したところ、同様の結論が導き出された。このXRPDパターンでは、エムトリシタビンの2θピーク位置もまた観察された。
【0081】
【表3A】
【表3B】
【0082】
マイクロリットルスケールでのTDFA2:1の結晶化
少量の、約2-3mgの市販の出発物質を、プレートウェルに仕込んだ。出発物質は、テトラヒドロフラン/水(80/20 v/v)混合物中に、ストック添加(stock-dose)した。溶媒を、20kPa未満で約45乃至75時間に亘る蒸発により除去し、出発物質を乾燥させた。結晶化溶媒または結晶化溶媒の混合物(50/50 v/v)を、乾燥出発物質を入れたウェルに少量ずつ室温にて加え、40mlの全容積及び50または80mg/mlのストック濃度とした。溶液を加熱し、60℃に30分間に亘って維持した。次いで、約1℃/時間または50℃/時間の冷却速度で、5℃または20℃の最終温度にまで制御冷却を行い、この温度に1、48、75、117、または139時間に亘って維持した。その後、溶媒を20kPaの圧力下、室温にて、48-120時間に亘って蒸発させた。得られた残渣を、X線粉末回折、DSC、及びTG-MSによって分析した。使用した溶媒は、表Iの通りである。HPLCバイアル中での特定の実験では、301.6mgの出発物質を2-メチル-4-ペンタノールに溶解させた。溶液を、60℃に30分間加熱した。次いで、約1℃/時間の冷却速度で室温(約22℃)にまで制御冷却を行い、この温度に48時間に亘って維持した。次いで、固形物質を遠心分離によって上澄み溶液から分離させた。固形物質のXRPD測定により、これがD形態であることが判明した。上澄み溶液を蒸発させ、残渣のXRPDによって、これがフマル酸及び少量のD形態であることが判明した。この実験により、1:1塩であるテノホビルDFから2:1共結晶であるTDFA2:1への転化の際に、過剰のフマル酸が確認される。
【0083】
熱時濾過を用いるミリリットルスケールでのTDFA2:1の結晶化
少量の、約70-75mgの市販の出発物質を、HPLCバイアルに仕込んだ。結晶化溶媒(または溶媒の50/50 v/v混合物)を、少量ずつ、乾燥出発物質を入れたバイアルに室温にて加え、200-1000マイクロリットルの全容積とした。用いた溶媒及び条件は、表IIの通りである。その後、溶液を、60分間について20乃至60℃の速度で加熱し、この温度で濾過した。濾液を1.1または50℃/時間で3または20℃に冷却し、この温度を24時間維持した。次いで、溶媒を、20kPaの圧力下で、20-25℃にて、15-200時間に亘ってバイアルから蒸発させた(表II参照、0.2kPaで500時間に亘る、(R)-(-)-2-オクタノールの場合)。得られた残渣を、X線粉末回折、DSC、及びTGAによって分析した。
【0084】
貧溶媒添加を用いるミリリットルスケールでのTDFA2:1の結晶化
貧溶媒添加実験を、2つの異なるプロトコルに従って行った。各溶媒についての第一プロトコル(正方向貧溶媒添加)により、スラリーを常温で調製し、これを約17-19時間に亘って平衡させた後に濾過してバイアルに入れた。貧溶媒を、1:1の溶媒:貧溶媒比を用いて添加した。この比は、沈降が全く起こらない場合には、引き続き貧溶媒を用いて1:4に増大させた。添加の時間間隔は1時間であった。貧溶媒の総容積は、飽和溶液の容積と同等であった。
【0085】
第二プロトコル(逆方向貧溶媒添加)としては、スラリーを常温で調製し、これを約17-19時間に亘って平衡させた後に濾過して4つのバイアルのセットに入れた。これらの各バイアルの内容物を、貧溶媒を入れたバイアルに加えた。飽和溶液の4つのバイアルの総容積は、貧溶媒の容積と同等であった。添加の時間間隔は1時間であった。
【0086】
沈降物を遠心分離によって回収し、固形生成物を乾燥させてXRPDによって分析した。沈降が全く起こらない場合には、溶液を96-314時間に亘って室温で蒸発させ、残渣をXRPDによって分析した。実験の詳細については、表IIIを参照のこと。
【0087】
スラリー結晶化を用いる、ミリリットルスケールでのTDFA2:1の結晶化
約50mgの出発物質を使用して、溶媒と共にスラリーを作製した(表4参照)。スラリーを、表4に示される通り、2及び10日間の時間間隔で室温または35℃にて撹拌した。この物質を、固形形態の変化を確認するためにXRPDによって検査した。同様のスラリー実験によって得られた物質のXRPDパターンにおいて、フマル酸の強度ピークが、約28.7度2θに観察されたが、これは、1:1テノホビルDFから2:1共結晶への転化の結果としての、スラリー物質中の過剰のフマル酸を示す。
【0088】
【表4】
【0089】
水浴中、室温及び35℃でのスラリー実験により、2日後には出発物質から形態TDFA2:1への添加がもたらされた。10日後のスラリー中の物質のXRPD測定により、固体形態が、依然として、形態TDFA2:1であることが判明した。出発物質を、1,4-ジオキサン及びアセトニトリル中とした、室温でのスラリー実験は、2日及び10日後にいかなる固形形態転化ももたらさなかった。
【0090】
シード結晶化を用いる、ミリリットルスケールでのTDFA2:1の結晶化
3つのタイプのシード実験を、下記の通り行った。
タイプ1
スラリーを、室温にて、約100mgの出発物質を用いて作製した。スラリーを室温で濾過し、少量の約2mgの対応するシードを添加した。溶液を、室温または5℃に、一晩維持した。次いで、溶液を蒸発させ、固形物質をXRPDによって検査した。
タイプ2
実験を、貧溶媒添加の例に記載した通りに、以下の変更と共に行った:沈降の直後に、少量の約2mgの対応するシードを、溶液に添加した。溶液を、室温または5℃に、一晩維持した。次いで、溶液を蒸発させ、固形物質をXRPDによって検査した。
タイプ3
スラリーを、室温にて、約100mgの出発物質を用いて作製した。少量の約5mgの対応するシードを添加した。スラリーを約1時間撹拌した後、これを2日間室温に維持した。次いで、溶液を蒸発させ、固形物質をXRPDによって検査した。
各実験で使用した具体的条件及びシードは、表5に示される。
【0091】
【表5】
【0092】
ミリリットルスケールでのTDFA2:1の結晶化
2,2,2トリフルオロエタノールから:
少量の、約15.8mgの出発物質を、HPLCバイアルに仕込んだ。2,2,2トリフルオロエタノールを、少量ずつ、乾燥出発物質を入れたバイアルに室温にて加え、1000マイクロリットルの全容積とした。バイアルを振盪し、溶解度を目視で評価した。溶液を加熱し、60℃に30分間に亘って維持した。次いで、溶媒を、減圧下で20-25℃にて、バイアルから蒸発させた。蒸発時間及び圧力は、20kPaにて22.5時間であった。蒸発を、4.4kPaにて71時間に亘って継続させた。得られた残渣を、X線粉末回折、DSC、及びTGAによって分析した。
【0093】
アセトンから:
少量の、約15.3mgの出発物質を、HPLCバイアルに仕込んだ。溶媒のアセトンを、少量ずつ、乾燥出発物質を入れたバイアルに室温にて加え、1000マイクロリットルの全容積とした。バイアルを振盪し、溶解度を目視で評価した。次いで、溶媒を、減圧下で20-25℃にて、バイアルから蒸発させた。蒸発時間及び圧力は、20kPaにて22.5時間であった。得られた残渣を、X線粉末回折、DSC、及びTGAによって分析し、TDFA2:1として同定した。
【0094】
ジクロロメタンから:
少量の、約12.4mgの出発物質を、HPLCバイアルに仕込んだ。溶媒のジクロロメタンを、少量ずつ、乾燥出発物質を入れたバイアルに室温にて加え、1000マイクロリットルの全容積とした。バイアルを振盪し、溶解度を目視で評価した。溶液を加熱し、60℃に30分間に亘って維持した。次いで、溶媒を、減圧下で20-25℃にて、バイアルから蒸発させた。蒸発時間及び圧力は、20kPaにて22.5時間であった。得られた残渣を、X線粉末回折、DSC、及びTGAによって分析し、テノホビルDF形態のTDFA2:1として同定した。
【0095】
ニトロメタンから:
少量の、約15.9mgの出発物質を、HPLCバイアルに仕込んだ。溶媒のニトロメタンを、少量ずつ、乾燥出発物質を入れたバイアルに室温にて加え、1000マイクロリットルの全容積とした。バイアルを振盪し、溶解度を目視で評価した。溶液を加熱し、60℃に30分間に亘って維持した。次いで、溶媒を、減圧下で20-25℃にて、バイアルから蒸発させた。蒸発時間及び圧力は、20kPaにて22.5時間であった。得られた残渣を、X線粉末回折、DSC、及びTGAによって分析し、テノホビルDF形態のTDFA2:1として同定した。
【0096】
水から:
少量の、約16.9mgの出発物質を、HPLCバイアルに仕込んだ。溶媒の水を、少量ずつ、乾燥出発物質を入れたバイアルに室温にて加え、1000マイクロリットルの全容積とした。バイアルを振盪し、溶解度を目視で評価した。溶液を加熱し、60℃に30分間に亘って維持した。次いで、溶媒を、減圧下で20-25℃にて、バイアルから蒸発させた。蒸発時間及び圧力は、20kPaにて22.5時間であった。蒸発を、4.4kPaにて71時間に亘って継続させた。得られた残渣を、X線粉末回折、DSC、及びTGAによって分析し、テノホビルDF形態のTDFA2:1として同定した。
【0097】
動的蒸気吸着(DVS)
水分吸着等温線を、Surface Measurement Systems(英国、ロンドン)社製のDVS-1システムを用いて測定した。様々な形態の固形物質の水分取込における相違により、増大する相対湿度についての様々な固形形態の相対安定性における相違が示される。実験は、25℃の一定温度で行った。
【0098】
約11.5mgの形態TDFA2:1の試料を、DVSパン内に広げた。試料を、相対湿度0%にて7時間に亘って乾燥させた。次いで、チャンバーの相対湿度を5%単位毎に0%から95%に増大させ、水蒸気の吸着を観察した。試料を、各段階に1時間に亘って維持した。次いで、相対湿度を5%単位毎に0%にまで低減させ、各段階に1時間に1時間維持することによって、吸着を観察した。吸着−脱離サイクルのグラフは、図3に示される。水蒸気の総取込は約0.8%であったが、これは吸湿性についての業界基準に合致する。購入したままの出発物質を用いる同様のDVS実験では、総蒸気取込は約4%であったが、これは製剤において望ましくなく、更なる対策を要する。実験の終わりに、固形物質をXRPDにより測定したところ、構造には全く変化のないことが判明した(図4)。
【0099】
実施例
TDFA 2:1とULT 1との薬物動態比較研究
TDFA 2:1及びULT 1のバッチを、μM篩いで篩うことによって同等の結晶サイズをもたせて調製した。小型のセルロースカプセルに、およそ15mgのテノホビルDF形態TDFA 2:1とテノホビルULT Yとのいずれかを充填した。それぞれおよそ300グラムの12匹のオスのWisterラットに、形態TDFA 2:1とテノホビルULT 1とのいずれかのカプセル1つを、経口チューブにより投与し、次いで1mlの水道水を投与した。各ラットから、一定の間隔をおいて、尾静脈の穿刺によって少量の血液を採取した。血液試料は、更なる処理のために、即座に液体窒素中で凍結させた。
【0100】
全ての試料を採取した後、各試料の血漿調製物を作製した。血漿試料を、LC-MS-MSによる分析のために、そのテノホビル含量について更に検査した。採取の効率性を、ラット血漿試料に既知量のテノホビルを混ぜることによる比較によって決定した。テノホビル(テノホビルDFの活性代謝物)の濃度を、較正曲線に対してLC-MS-MSを用いて、各試料において定量した。薬物動態の比較結果を、図5に示す。PKデータから、テノホビルジソプロキシルのヘミフマレート共結晶であるTDFA 2:1は、市販のテノホビルジソプロキシルのフマル酸塩であるテノホビルジソプロキシル形態ULT 1と、生物学的に同等であると結論づけられた。
【0101】
表3:テノホビルDF形態TDFA 2:1の最終座標及び等価等方変位
【表6A】
【表6B】
【表6C】
【表6D】
【0102】
【表7】
【0103】
【表8】
【0104】
【表9】
【技術分野】
【0001】
本発明は、テノホビルジソプロキシルとフマル酸との、モル比2:1での新規な共結晶組成物、その調製方法、及びその形成、並びに医薬品分野、特に抗ウィルス医薬品の分野における応用に関する。
【背景技術】
【0002】
テノホビルジソプロキシルフマレート(DF)は、米国において、単独でまたは別の抗レトロウィルス薬と組み合わせてHIV-I感染の治療用に認可された、ヌクレオチド系逆転写酵素阻害剤である。テノホビルジソプロキシルDFは、商品名VIREAD(登録商標)(Gilead Science, Inc.)として市販されており、別の抗ウィルス剤との組み合わせとしてTRUVADA(登録商標)及びATRIPLA(登録商標)抗HIV薬剤として市販されている。
【0003】
開発されている抗HIV薬剤の中には、HIV逆転写酵素(RT)またはプロテアーゼ酵素をターゲットとするものがあるが、これらの酵素はいずれもウィルスの複製に必要とされるものである。RT阻害剤の例には、ヌクレオシド/ヌクレオチドRT阻害剤(NRTI類)及び非ヌクレオチド系RT阻害剤(NNRTI類)が含まれる。現在、HIV感染患者は、3つの複合薬で規定通り治療されている。(少なくとも)3つのNRTI類を含む投薬計画、2つのNRTI類と1つまたは2つのプロテアーゼ阻害剤(PI)類との複合薬を含む投薬計画、あるいは2つのNRTI類と1つのNNRTIとの複合薬を含む投薬計画が、広く用いられている。これらの複合薬において2つ以上のPI類が使用される場合には、PI類の1つはしばしばリトナビルであり、これは治療量以下の低い用量で与えられ、投薬計画内の別のPI類の除去の効果的な阻害剤として作用して、ウィルスの最大限の抑制をもたらし、これにより耐性発現を低減する。
【0004】
臨床研究により、これらの抗HIV薬剤の3剤複合薬は、疾病進行及び死の回避に、1つの薬剤を単独でまたは2剤複合薬を用いるよりも格段に効果的であることが判明している。こうした薬剤の様々な複合薬を用いた、薬剤複合物の多数の研究により、こうした複合薬によってHIVに感染した人々において疾病進行及び死が大幅に低減されることが確立している。現在一般的に抗HIV薬剤の複合薬に与えられている名称は、HAART(高活性レトロウィルス療法)である。
【0005】
テノホビルDF、テノホビルジソプロキシル、TDF、ビス-POC-PMPAとしても既知である(米国特許第5,935,946号明細書、第5,922,695号明細書、第5,977,089号明細書、第6,043,230号明細書、第6,069,249号明細書)、テノホビルジソプロキシルフマレートは、テノホビルのプロドラッグ塩である。テノホビルジソプロキシルフマレートの化学名は、9-[(R)-2-[[ビス[[(イソプロポキシカルボニル)オキシ]メトキシ]ホスフィニル]-メトキシ]プロピル]アデニンフマレート(1:1)である。CAS登録番号は、202138-50-9である。これは、C19H30N5O10P・C4H4O4の分子式及び635.52の分子量を有する。これは以下の構造式:
【化1】
を有する。
【0006】
テノホビルDFの結晶形態は、とりわけ、国際特許公報第99/05150号、欧州特許第998480号明細書、及び米国特許第5935946号明細書に記載されている。この結晶形態(Gilead 1)は、約4.9、10.2、10.5、18.2、20.0、21.9、24.0、25.0、25.5、27.8、30.1、及び30.4のXRPDピークを有することにより特徴付けられる。更にまた、これらの結晶は、不透明または乳白色であって、開始温度が約116℃である、約118℃でのDSC吸収ピークを示し、且つそのIRスペクトルは、カイザーで表される、およそ3224、3107-3052、2986-2939、1759、1678、1620、1269、及び1102に特性帯を示すと記載されている。嵩密度は、約0.15-0.30g/mL、通常は約0.2-0.25g/mLと記載されている。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】米国特許第5,935,946号明細書
【特許文献2】米国特許第5,922,695号明細書
【特許文献3】米国特許第5,977,089号明細書
【特許文献4】米国特許第6,043,230号明細書
【特許文献5】米国特許第6,069,249号明細書
【特許文献6】国際特許公報第99/05150号
【特許文献7】欧州特許第998480号明細書
【特許文献8】米国特許第5935946号明細書
【発明の概要】
【発明が解決しようとする課題】
【0008】
テノホビルDFを含む幾つかの市販製品を分析したところ、これらが、固体形態のテノホビルDFの様々な割合の混合物を含むことが判明した。本発明者により、市販製品中の固体形態のテノホビルDFが、一般的に少なくとも2つの形態の混合物である印が発見されている。これらの形態の1つが、応力を受けた場合、例えば高温下及び/または高湿度下におかれた場合、その結晶形態から別の形態への転化を経ることもまた判明している。本発明者は、水の存在が1つの形態から別の形態への転化を誘発または促進すると考えている。このことは、市販製品中に現在使用されている固体形態が安定でないか、または少なくとも低い安定性を有することを示唆する。市販製品中におけるテノホビルジソプロキシルのフマル酸に対するモル容積比は、一般的に1:1とされる。
【課題を解決するための手段】
【0009】
本発明は、テノホビルジソプロキシルとフマル酸との、モル比2:1での新規な共結晶(TDFA 2:1)に関する。本発明は、1:1のフマレート塩である、テノホビルDFとは異なる。本発明のTDFA 2:1共結晶は、現在既知であるテノホビルDFの結晶形態(Gilead 1)よりも安定性が高く、且つ吸湿性が低い。
る。
【図面の簡単な説明】
【0010】
【図1A】図1Aは、テノホビルジソプロキシルとフマル酸との2:1モル比での共結晶(TDFA 2:1)のX線粉末回折パターンを示す図である。
【図1B】図1Bは、テノホビルジソプロキシルとフマル酸との2:1モル比での共結晶(TDFA 2:1)のDSCサーモグラムを示す図である。
【図1C】図1Cは、テノホビルジソプロキシルとフマル酸との2:1モル比での共結晶(TDFA 2:1)のTGAサーモグラムを示す図である。
【図1D】図1Dは、単結晶のデータから測定される、テノホビルジソプロキシルとフマル酸との2:1モル比での共結晶(TDFA 2:1)由来の遊離塩基−遊離酸−遊離塩基体の分子構造を示す図である。
【図1E】図1Eは、テノホビルジソプロキシルとフマル酸との2:1モル比での共結晶(TDFA 2:1)の結晶充填を示す図である。
【図1F】図1Fは、テノホビルジソプロキシルとフマル酸との2:1モル比での共結晶(TDFA 2:1)のラマンスペクトルを示す図である。
【図2A】図2Aは、粉砕したViread錠剤から得られるX線粉末回折パターンを示す図である。
【図2B】図2Bは、コーティングを除去したVireadの錠剤から得られるX線粉末回折パターンを示す図である。
【図2C】図2Cは、粉砕したTruvada錠剤から得られたX線粉末回折パターンを示す図である。
【図3】図3は、TDFA 2:1形態の吸着(ダイヤ形)及び脱離(四角形)挙動のDVSプロットを示す図である。
【図4】図4は、DVS測定前(上)及びDVS測定後(下)TDFA 2:1の実験的XRPDパターンを示す図である。
【図5】図5は、TDFAの生物学的同等性を示す、薬物動態データである。
【発明を実施するための形態】
【0011】
テノホビルジソプロキシル/フマル酸 共結晶(TDFA 2:1)
本発明は、2単位のテノホビルジソプロキシルが、1単位のフマル酸と、実験式C19H30N5O10P・C4H4O4をもって共結晶化した、テノホビルジソプロキシルとフマレートとの共結晶に関する。この共結晶は、テノホビルジソプロキシルのヘミフマル酸共結晶である。1つの態様では、本発明は、実質的に純粋な組成物、特にテノホビルジソプロキシルとフマル酸との、モル比2:1での共結晶(TDFA 2:1)を提供する。共結晶とは、1つまたは複数の分子性物質が、単位格子内に取り込まれた結晶体である。共結晶には、通常、塩類、例えばテノホビルDFが含まれず、こうした塩類は、逆帯電したイオン間及び溶媒和物間の静電結合をもたらすプロトン移動によって識別され、その結合機構は共結晶におけるものと同様であってもよいが、これらはその結晶化する際の溶媒と基質との会合である。例えば、Visheweshwar, P.; McMahon, J. A.; Bis, J. A.; Zaworotko, M. J. (2006) J. Pharm. Sci. 95(3), 499-516を参照のこと。
【0012】
上述の通り、本発明の新規な固体形態のTDFA 2:1は、独立して、実質的に純粋形態であり、好ましくは実質的に別のアモルファス及び/または結晶固体形態、例えば前述の従来技術に記載されている固体形態、すなわち本明細書中の別の箇所にも記載されるGilead 1またはULT-1を含まない。この点について、「実質的に含まない」とは、少なくとも約80%、85%、90%、95%、96%、97%、98%、または99%の純粋化合物に関する。この点について、「実質的に別のアモルファス及び/または結晶固体形態を含まない」とは、本発明による形態中に存在する、これらの別のアモルファス及び/または結晶固体形態が、約20%、15%、10%、5%、4%、3%、2%、1%以下であることを意味する。
【0013】
本発明の共結晶は、室温未満の温度、好ましくは120K前後の温度で共結晶である。本発明の共結晶は、より高い温度、例えば室温でも共結晶である。室温での単結晶構造及び実験的XRPDパターンにより、120Kと室温との間では構造相転移が全く起こっておらず、これらの温度でのXRPDパターンにおける相違は熱膨張によることが示される。以上に基づいて、TDFA 2:1は、室温(測定の地理的な位置によって15乃至40℃)では共結晶である。
【0014】
TDFA 2:1は、7.9、9.8、11.0、12.0、13.7、14.3、16.1、16.8、18.0、19.2、20.4、21.2、21.7、22.6、23.4、24.3、25.4、27.6度2θ±0.3度2θ、好ましくは±約0.2度2θ、更に好ましくは±約0.1度2θ、最も好ましくは±約0.05度2θからなる群より選択される、少なくとも1つ、好ましくは少なくとも2つ、より好ましくは少なくとも3つ、更にいっそう好ましくは少なくとも4つ、特に好ましくは少なくとも5つ、最も好ましくは6つのX線粉末回折ピークの選択によって特徴付けられる。好ましい実施態様では、少なくとも7つ、より好ましくは少なくとも8つ、更にいっそう好ましくは少なくとも9つ、特に好ましくは少なくとも10、最も好ましくは11のX線粉末回折ピークが上記群より選択される。より好ましい実施態様では、少なくとも12、より好ましくは少なくとも13、更にいっそう好ましくは少なくとも14、特に好ましくは少なくとも15、最も好ましくは16のX線粉末回折ピークが上記群より選択される。
【0015】
好ましくは、TDFAは、7.82、8.09、11.95、16.80、21.20、22.52、24.29度2θからなる群より選択される、少なくとも1つ、好ましくは少なくとも2つ、より好ましくは少なくとも3つ、更にいっそう好ましくは少なくとも4つ、特に好ましくは少なくとも5つ、最も好ましくは6つのX線粉末回折ピークの選択によって特徴付けられる。2θの位置は、室温で、1.54178Åの波長を用いて、TDFA 2:1の単結晶構造から算出される。実験的XRPDパターンにおいては、実験設定及びピーク重なりにより、以上に挙げた値から偏差があってもよい。
【0016】
TDFA 2:1は、下記のX線回折ピーク群によって、さらに任意に付随する強度によって、特徴付けることができる。
【表1】
【0017】
別の実施態様では、TDFA 2:1は、実質的に図1Aに従う、X線粉末回折パターンによって特徴付けられる。
別の実施態様では、TDFA 2:1は、実質的に図1Bに従う、DSCによって特徴付けられる。
別の実施態様では、TDFA 2:1形態は、実質的に図1Cに従う、TGAによって特徴付けられる。
別の実施態様では、本発明のTDFA 2:1形態は、開始温度が105.3℃である、117.0℃での特徴ピークを示すDSCによって特徴付けられるが実質的に図1Bに従う、DSCによって特徴付けられる。熱分析により、共結晶TDFA 2:1は非溶媒和であると結論づけられる。
【0018】
本発明は、一態様において、表Iに記載される適当な溶媒またはその混合物に、テノホビルDFを溶解させるかまたは混入し、溶媒の蒸発によりTDFA 2:1を結晶化させる工程を含む、共結晶TDFA 2:1の調製方法に関する。
【0019】
本発明は、別の態様において、表IIに記載される適当な溶媒またはその混合物に、テノホビルDFを溶解させるかまたは混入し、飽和溶液の冷却及び/または蒸発結晶化によりTDFA 2:1を結晶化させる工程を含む、共結晶TDFA 2:1の調製方法に関する。
【0020】
本発明は、一態様において、表IIIに記載される適当な溶媒またはその混合物に、テノホビルDFを溶解させるかまたは混入し、表IIIに記載される貧溶媒添加によりTDFA 2:1を結晶化させる工程を含む、共結晶TDFA 2:1の調製方法に関する。
【0021】
本発明は、別の態様において、本明細書中の別の箇所(溶媒に関する章)に概説される適当な溶媒またはその混合物にテノホビルDFを溶解させるかまたは混入し、スラリー結晶化及び/またはシード結晶化によりTDFA 2:1を結晶化させる工程を含む、共結晶TDFA 2:1の調製方法に関する。
【0022】
本発明の共結晶は、図1D及び/または1Eに図示され、且つ/または表2及び3に記載されるTDFA 2:1の単結晶構造に関する一態様によっても特徴付けられる。
【0023】
【表2】
【0024】
一態様では、本発明はさらに、(参照のために援用することとする、出願人による同時係属出願である米国第60/873,267号明細書に記載の通り、)実質的に純粋であり、好ましくはテノホビルDF形態のULT-1を含まないTDFA 2:1に関する。米国第60/873,267号明細書に記載のテノホビルDF形態のULT-1は、5.0、5.5、10.3、10.6、10.9、11.4、14.2、17.3、18.3、19.9、22.0、22.9、25.0、27.9、30.1度2θ±0.3度2θ、好ましくは±約0.2度2θ、更に好ましくは±約0.1度2θ、最も好ましくは±約0.05度2θからなる群より選択される、少なくとも1つ、好ましくは少なくとも2つ、より好ましくは少なくとも3つ、更にいっそう好ましくは少なくとも4つ、特に好ましくは少なくとも5つ、最も好ましくは6つのX線粉末回折ピークの選択によって特徴付けられる。好ましい実施態様では、少なくとも7つ、より好ましくは少なくとも8つ、更にいっそう好ましくは少なくとも9つ、特に好ましくは少なくとも10、最も好ましくは11のX線粉末回折ピークが上記群より選択される。より好ましい実施態様では、少なくとも12、より好ましくは少なくとも13、更にいっそう好ましくは少なくとも14、特に好ましくは少なくとも15のX線粉末回折ピークが上記群より選択される。
【0025】
本発明の好ましい実施態様では、TDFA 2:1は、固体形態であるテノホビルDF形態のULT-1を実質的に含まない。本発明の好ましい実施態様では、TDFA 2:1は、5.0及び/または5.5度2θ±0.1度2θにX線ピークを有することによって特徴付けられる固体形態を、実質的に含まない。別の好ましい実施態様では、TDFA 2:1は、4.9及び/または5.4度2θ±0.1度2θにX線ピークを有することによって特徴付けられる固体形態を、実質的に含まない。別の好ましい実施態様では、TDFA 2:1は、4.97及び/または5.44度2θ±0.1度2θにX線ピークを有することによって特徴付けられる固体形態を、実質的に含まない。
【0026】
一態様では、本発明は、実質的に純粋であり、好ましくは、(本明細書中の別の箇所及び出願人による同時係属出願である米国第60/873,267号明細書に記載の通り)テノホビルDF形態のULT-1から得られるTDFA 2:1形態を含む、医薬品組成物に関する。
【0027】
一態様では、本発明は、Cipla社より入手される出発物質テノホビルDFから、表I、II、及び/またはIIIの1つまたは複数に挙げられた有機溶媒、あるいはこれらの混合物からの再結晶により、2:1ヘミフマル酸共結晶を生成させる、TDFA 2:1形態の調製方法に関する。
【0028】
一態様では、本発明は、水性雰囲気における結晶化による、テノホビルDF形態からのTDFA 2:1の調製方法に関する。
【0029】
共結晶の単結晶が、水、メタノール、イソプロピルアセテート、(R)-(-)-2-オクタノール中のテノホビルDFの飽和溶液の、室温またはより低温、好ましくは5℃での緩慢蒸発によって得られた。別の実施態様では、飽和溶液を1℃/時乃至5℃/時の冷却速度で冷却した後、この温度に数日おく。表I、II、及びIIIに挙げた溶媒から、共結晶TDFA 2:1を得ることも可能である。
【0030】
溶媒
本発明のTDFA 2:1の調製方法の所定の実施態様では、蒸発結晶化、熱時濾過貧溶媒添加、シード結晶化及び/またはスラリー結晶化のための溶媒は、好ましくは、(R)-(-)-2-オクタノール、1,2-ジエトキシエタン、1,2-ジメトキシエタン、1,4-ジオキサン、1-ブタノール、1-ヘプタノール、1-ヘキサノール、1-メトキシ-2-プロパノール、1-ニトロプロパン、1-オクタノール、2,2,2-トリフルオロエタノール、2-ブタノン、2-エトキシエタノール、2-エトキシエチルアセテート、2-ヘキサノール、2-メトキシエタノール、2-ニトロプロパン、2-ペンタノール、2-プロパノール、4-ヒドロキシ-4-メチル-2-ペンタノン、アセトン、アセトニトリル、ブチロニトリル、シクロヘキサノール、シクロペンタノール、シクロペンタノン、ジエチレングリコールジメチルエーテル、ジメチルカーボネート、ジメチルカーボネート、エタノール、エチルホルメート、エチルアセテート、エチレングリコールモノブチルエーテル、ジクロロメタン、フルフリルアルコール、イソブタノール、イソプロピルアセテート、メタノール、メトキシエチルアセテート、メチルアセテート、メチルブチレート、メチルプロピオネート、2-メチル-4-ペンタノール、N,N-ジメチルアセトアミド、N,N-ジメチルホルムアミド、ニトロベンゼン、ニトロエタン、ニトロメタン、N-メチルピロリドン、プロピオニトリル、プロピルアセテート、プロピレングリコールメチルエーテルアセテート、tert-ブタノール、テトラヒドロフラン、テトラヒドロフルフリルアルコール、テトラヒドロピラン、水、及びこれらの混合物から成る群より選択される。
【0031】
本発明のTDFA 2:1の調製方法の所定の実施態様では、蒸発結晶化、熱時濾過貧溶媒添加、シード結晶化及び/またはスラリー結晶化のための溶媒は、更に好ましくは、(R)-(-)-2-オクタノール、1,2-ジエトキシエタン、1,2-ジメトキシエタン、1,4-ジオキサン、1-ブタノール、1-ニトロプロパン、1-プロパノール、2-ブタノン、2-エトキシエチルアセテート、2-メチル-4-ペンタノール、2-ニトロプロパン、2-プロパノール、アセトン、アセトニトリル、シクロペンタノール、エタノール、イソブタノール、イソプロピルアセテート、メタノール、メトキシ-2-1-プロパノール、メチルプロピオネート、N,N-ジメチルアセトアミド、N,N-ジメチルホルムアミド、ニトロメタン、tert-ブタノール、テトラヒドロフラン、水、1,2-ジクロロエタン、2,6-ジメチル-4-ヘプタノン、アミルエーテル、ブチルベンゼン、クロロホルム、ジクロロメタン、ヘキサフルオロベンゼン、n-ヘプタン、N-メチルピロリドン、tert-ブチルメチルエーテル、トルエン、シクロペンタノンから成る群より選択される。
【0032】
本発明のTDFA 2:1の調製方法の所定の実施態様では、熱時濾過結晶化のための溶媒は、好ましくは、(R)-(-)-2-オクタノール、1,2-ジエトキシエタン、1,2-ジメトキシエタン、1,4-ジオキサン、1-ブタノール、1-ニトロプロパン、1-プロパノール、2-ブタノン、2-エトキシエチルアセテート、2-メチル-4-ペンタノール、2-ニトロプロパン、2-プロパノール、アセトン、アセトニトリル、シクロペンタノール、エタノール、イソブタノール、イソプロピルアセテート、メタノール、メトキシ-2-1-プロパノール、メチルプロピオネート、N,N-ジメチルアセトアミド、N,N-ジメチルホルムアミド、ニトロメタン、tert-ブタノール、テトラヒドロフラン、水、及びこれらの混合物から成る群より選択される。
【0033】
本発明のTDFA 2:1の調製方法の所定の実施態様では、溶媒/貧溶媒結晶化のための溶媒は、好ましくは、1,2-ジクロロエタン、1,2-ジメトキシエタン、1,4-ジオキサン、2,6-ジメチル-4-ヘプタノン、2-ブタノン、アセトン、アセトニトリル、アミルエーテル、ブチルベンゼン、クロロホルム、シクロヘキサン、シクロヘキサン、ジクロロメタン、ヘキサフルオロベンゼン、メタノール、n-ヘプタン、ニトロメタン、N-メチルピロリドン、tert-ブチルメチルエーテル、テトラヒドロフラン、トルエン、水、及びこれらの混合物から成る群より選択される。
【0034】
本発明のTDFA 2:1の調製方法の所定の実施態様では、貧溶媒結晶化のための貧溶媒は、好ましくは、1,2-ジクロロエタン、2,6-ジメチル-4-ヘプタノン、アセトン、アミルエーテル、ブチルベンゼン、クロロホルム、シクロヘキサン、ジクロロメタン、ヘキサフルオロベンゼン、n-ヘプタン、ニトロメタン、tert-ブチルメチルエーテル、トルエン、及びこれらの混合物から成る群より選択される。
【0035】
本発明のTDFA 2:1の調製方法の所定の実施態様では、シード結晶化のための溶媒は、好ましくは、メタノール、水、1,4-ジオキサン、アセトニトリル、2-エトキシエチルアセテート、2-メチル-4-ペンタノール、テトラヒドロフラン、ブチルベンゼン、アミルエーテル、tert-ブチルメチルエーテル、シクロペンタノン、及びこれらの混合物から成る群より選択される。
【0036】
本発明のTDFA 2:1の調製方法の所定の実施態様では、スラリー結晶化のための溶媒は、好ましくは、水、メタノール、アセトニトリル、1,4-ジオキサン、及びこれらの混合物から成る群より選択される。
【0037】
医薬品製剤
本発明は、更に、テノホビルDFの新規な結晶形態を含む医薬品製剤に関する。
本発明の医薬品製剤は、本明細書中に開示されるTDFA 2:1を含む。本発明はまた、本発明による結晶形態を一つまたは複数含む医薬品組成物を提供する。本発明の製剤処方は、本発明による結晶形態の一つまたは複数を活性成分として、任意に一つまたは複数の別の結晶形態との混合物として含む。
【0038】
本発明による製剤処方は、TDFA 2:1形態に加えて、付加的な医薬品活性成分、好ましくは、抗HIV剤、更に好ましくは、エファビレンツ、エムトリシタビン、リトナビル、及び/またはTMC114をさらに含んでよい。
【0039】
活性成分に加えて、本発明の医薬品製剤は、一つまたは複数の賦形剤を含んでよい。賦形剤は、様々な目的で製剤に加えられる。
【0040】
希釈剤は、固形医薬品組成物の体積を増大させ、患者及び介護人にとってのこの組成物を含む医薬品投薬形態の取り扱いを、より容易にしうる。固形組成物の希釈剤には、例えば、マイクロクリスタリンセルロース(例えばAvicel(登録商標))、マイクロファインセルロース、ラクトース、澱粉、アルファ化澱粉、炭酸カルシウム、硫酸カルシウム、糖、デキストレート類、デキストリン、デキストロース、リン酸水素カルシウム二水和物、リン酸三カルシウム、カオリン、炭酸マグネシウム、酸化マグネシウム、マルトデキストリン、マンニトール、ポリメタクリレート類(例えばEudragit(登録商標))、塩化カリウム、粉末セルロース、塩化ナトリウム、ソルビトール、及びタルクが含まれる。
【0041】
投薬形態、例えば錠剤にコンパクト化された固形医薬品組成物は、その機能の中に、圧縮後に活性成分と別の賦形剤との互いとの結合を補助することを含む、賦形剤を含んでよい。固形医薬品組成物のための結合剤には、アカシア、アルギン酸、カーボマー(例えば、Carbopol)、カルボキシメチルセルロースナトリウム、デキストリン、エチルセルロース、ゼラチン、グアーガム、水添植物油、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース(例えば、Klucel(登録商標))、ヒドロキシプロピルメチルセルロース(例えば、Methocel(登録商標))、液体グルコース、ケイ酸アルミニウム・マグネシウム、マルトデキストリン、メチルセルロース、ポリメタクリレート類、ポビドン(例えば、Kollidon(登録商標)、Plasdone(登録商標))、アルファ化澱粉、アルギン酸ナトリウム、及び澱粉が含まれる。
【0042】
患者の胃の中のコンパクト化固形医薬品組成物の溶解速度は、この組成物への崩壊剤の添加によって増大されうる。崩壊剤には、アルギン酸、カルボキシメチルセルロースカルシウム、カルボキシメチルセルロースナトリウム(例えば、Ac-Di-Sol(登録商標)、Primellose(登録商標))、コロイド状二酸化ケイ素、クロスカルメロースナトリウム、クロスポビドン(例えば、Kollidon(登録商標)、Polyplasdone(登録商標))、グアーガム、ケイ酸アルミニウム・マグネシウム、メチルセルロース、マイクロクリスタリンセルロース、ポラクリリンカリウム、粉末セルロース、アルファ化澱粉、アルギン酸ナトリウム、澱粉グリコール酸ナトリウム(例えば、Explotab(登録商標))、及び澱粉が含まれる。
【0043】
流動促進剤は、非コンパクト化固形組成物の流動性を改善し、且つ投薬の正確性を改善するために加えることができる。流動促進剤として機能しうる賦形剤には、コロイド状二酸化ケイ素、三ケイ酸マグネシウム、粉末セルロース、澱粉、タルク、及びリン酸三カルシウムが含まれる。
【0044】
投薬形態、例えば錠剤が粉末組成物のコンパクト化によって製造される場合、この組成物には、パンチ及びダイから圧力がかけられる。賦形剤及び活性成分の中には、パンチ及びダイの表面に付着する傾向を有するものがあり、このため製品に孔が生じ、また別の表面凹凸が生じうる。滑剤を前記組成物に加えて、粘着性を低減し、製品のダイからの除去を容易にすることもできる。滑剤には、マグネシウムステアレート、カルシウムステアレート、グリセリルモノステアレート、グリセリルパルミトステアレート、水添蓖麻子油、水添植物油、鉱物油、ポリエチレングリコール、ナトリウムベンゾエート、ナトリウムラウリルスルフェート、ナトリウムステアリルフマレート、ステアリン酸、タルク、及び亜鉛ステアレートが含まれる。香味料及び調味料は、投薬形態を、患者にとってより口当たりの良いものにする。本発明の組成物に導入してもよい、医薬品向けの通常の香味料及び調味料には、マルトール、バニリン、エチルバニリン、メントール、クエン酸、フマル酸、エチルマルトール、及び酒石酸が含まれる。固形及び液状の組成物はまた、その外観を改善し、且つ/または患者による製品及び単位投与量レベルの確認を容易にするために、医薬品として許容されるいかなる着色剤を使用して染色してもよい。
【0045】
本発明の液状の医薬品組成物中では、本発明による結晶形態及び別のあらゆる固形賦形剤が、液状担体、例えば、水、植物油、アルコール、ポリエチレングリコール、プロピレングリコール、グリセリン、またはこれらの混合物中に、本明細書中に記載の結晶形態が維持される、すなわち溶解しない限りは、懸濁している。
【0046】
液状医薬品組成物は、乳化剤を含んで、液状担体に溶解しない活性成分または別の賦形剤を、この組成物全体に均一に分散させてよい。本発明の液状組成物中に使用してよい乳化剤には、例えば、ゼラチン、卵黄、カゼイン、コレステロール、アカシア、トラガカンス、ツノマタ、ペクチン、メチルセルロース、カーボマー、セトステアリルアルコール、及びセチルアルコールが含まれる。
【0047】
本発明の液状医薬品組成物は、増粘剤をさらに含んで、製品の口当たりを改善するか、且つ/または胃腸管の内壁を覆ってよい。こうした作用剤には、アカシア、アルギン酸ベントナイト、カーボマー、カルボキシメチルセルロースカルシウムもしくはナトリウム、セトステアリルアルコール、メチルセルロース、エチルセルロース、ゼラチングアーガム、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、マルトデキストリン、ポリビニルアルコール、ポビドン、プロピレンカーボネート、プロピレングリコールアルギネート、ナトリウムアルギネート、ナトリウム澱粉グリコレート、澱粉トラガカンス、及びキサンタンガムが含まれる。
【0048】
甘味料、例えば、ソルビトール、サッカリン、ナトリウムサッカリン、スクロース、アスパルテーム、フルクトース、マンニトール、及び転化糖を加えて、味を改善してよい。保存料及びキレート剤、例えば、アルコール、ナトリウムベンゾエート、ブチル化ヒドロキシルトルエン、ブチル化ヒドロキシアニソール、及びエチレンジアミンテトラ酢酸を、摂取に安全なレベルで加えて、貯蔵安定性を改善してもよい。本発明によれば、液状組成物は、グルコン酸、乳酸、クエン酸、または酢酸、ナトリウムグルコネート、ナトリウムラクテート、ナトリウムシトレート、またはナトリウムアセテートをさらに含んでよい。使用される賦形剤及びその量の選択は、経験及び標準的操作を勘案し、且つ当該分野の参考文献に基づいて、製剤研究者によって容易に決定されうる。
【0049】
眼または別の外部組織、例えば、口及び皮膚の感染のためには、この製剤は、好ましくは、活性成分を、例えば、0.01乃至10質量%の量で含む(0.1質量%刻みで0.1乃至5質量%の範囲で、例えば0.6質量%、0.7質量%の活性成分を含む)、好ましくは0.2乃至3質量%、最も好ましくは2質量%の量で含む、局所用軟膏またはクリームとして適用される。軟膏として製剤化される場合には、活性成分を、パラフィン系または水混和性のいずれの軟膏ベースと共に用いてもよい。
【0050】
あるいはまた、活性成分を、水中油型クリームベースを用いてクリーム中に製剤化してよい。
【0051】
所望により、クリームベースの水性相は、例えば、少なくとも30質量%の多価アルコール、すなわち、二つ以上のヒドロキシル基を有するアルコール、例えば、プロピレングリコール、ブタン1,3-ジオール、マンニトール、ソルビトール、グリセロール、及びポリエチレングリコール(PEG 400を含む)、及びこれらの混合物を含んでよい。局所用製剤は、望ましくは、皮膚または別の罹患した領域からの、活性成分の吸収または浸透を促進する化合物を含んでよい。こうした皮膚透過促進剤には、ジメチルスルホキシド及び関連類縁体が含まれる。
【0052】
本発明のエマルションの油性相は、既知の成分から既知の方法で構成されてよい。この相は一つの乳化剤(エマルジェントとしても既知)のみを含んでよい一方で、望ましくは、少なくとも一つの乳化剤の、脂肪または油、あるいは脂肪及び油の両方との混合物を含む。好ましくは、親水性乳化剤が、安定化剤として作用する親油性乳化剤と共に導入される。油と脂肪とをいずれも導入することもまた好ましい。安定化剤と共にまたは安定化剤なしに、乳化剤は乳化ワックスを構成し、ワックスと油及び脂肪とは、共に乳化軟膏ベースを構成するが、これはクリーム製剤の油性分散相を形成する。
【0053】
本発明の製剤中における使用に適当なエマルジェント及びエマルション安定化剤には、Tween8 60、Spans 80、セトステアリルアルコール、ベンジルアルコール、ミリスチルアルコール、グリセリルモノステアレート、及びナトリウムラウリルスルフェートが含まれる。
【0054】
前記製剤に適当な油または脂肪の選択は、所望の化粧品特性の達成に基づく。然るに、クリームは、好ましくは、チューブまたは別の容器からの漏出をさけるために適当なコンシステンシーを有する、非脂肪性、非着色性、及び洗浄可能な製品であるべきである。
【0055】
直鎖状または分枝状である、一塩基性もしくは二塩基性のアルキルエステル類、例えば、ジイソアジペート、イソセチルステアレート、ココナッツ脂肪酸類のプロピレングリコールジエステル、イソプロピルミリステート、デシルオレエート、イソプロピルパルミテート、ブチルステアレート、2-エチルヘキシルパルミテート、または、Crodamol CAPとして既知の分枝鎖エステル類のブレンドを使用してよく、最後の3つが好ましいエステル類である。これらは、要求される特性に応じて、単独でまたは組合せとして使用してよい。あるいはまた、高融点脂質、例えば白色軟質パラフィン及び/または流動パラフィン、あるいは別の鉱物油が使用可能である。
【0056】
眼への局所適用に適当な製剤には、活性成分が適当な担体中、特に、前記活性成分のための水性溶媒中に、溶解または懸濁された、点眼薬がさらに含まれる。活性成分は、適切には、こうした製剤中に、0.01乃至20質量%の濃度で存在し、0.1乃至10質量%である態様もあれば、約1.0質量%である態様もある。
【0057】
口への局所適用に適当な製剤には、調味ベース中、通常は、スクロース及びアカシアまたはトラガカンス中に、活性成分を含むロゼンジ類;不活性ベース中、例えば、ゼラチンとグリセリン、またはスクロースとアカシア中に、活性成分を含むトローチ類;及び適当な液状担体中に活性成分を含むマウスウォッシュ類が含まれる。
【0058】
直腸投与のための製剤は、例えば、ココアバターまたはサリチレートを含む適当なベースと共に、坐薬として提示してよい。
【0059】
担体が固形である、経鼻または吸入投与に適当な製剤には、例えば、1乃至500ミクロンの範囲の粒径(5ミクロン毎に20乃至500ミクロンの範囲、例えば30ミクロン、35ミクロン等の粒径を含む)を有する粉末が含まれる。担体が液状であって、経鼻スプレーまたは点鼻薬に適当な製剤には、活性成分の水性または油性の溶液が含まれる。
【0060】
エアロゾル投与に適当な製剤は、定法により調製してよく、別の治療剤と共に送達されてよい。吸入療法剤は、定量吸入器によって容易に投与される。
【0061】
膣内投与に適当な製剤は、活性成分に加えて、当業者に適当であることが知られた担体を含む、ペッサリー類、タンポン類、クリーム類、ゲル類、ペースト類、フォーム類、またはスプレー製剤として提示してよい。
【0062】
本発明の固形組成物には、粉末、粒子、凝集体、及びコンパクト化組成物が含まれる。用量には、経口、口腔、直腸、非経口(皮下、筋内、及び静脈内を含む)、吸入、眼科投与に適当な用量が含まれる。所与のあらゆる場合において、最適な投与は、治療しようとする状態の性質及び重篤度に依存するが、本集め胃の最も好ましい経路は経口である。用量は、単位用量形態で従来提示されているものでよく、医薬品分野において周知のいかなる方法で調製してもよい。
【0063】
投与形態には、錠剤、粉末、カプセル、坐薬、サシェ、トローチ、及びロゼンジ、ならびに液状シロップ、懸濁液、及びエリキシル剤が含まれる。
【0064】
本発明の投与形態には、前記組成物を、好ましくは粉末化もしくは粒状化した本発明の固形組成物を、硬質または軟質シェル内に含むカプセルであってよい。このシェルは、ゼラチン製であってよく、任意に、グリセリン及びソルビトール等の可塑剤及び乳白剤または着色剤を含む。
【0065】
活性成分及び賦形剤は、当業者に既知の方法によって、組成物及び投薬形態に製剤化してよい。錠剤化またはカプセル充填のための組成物は、湿式造粒法によって調製してよい。湿式造粒法では、粉末形態の活性成分及び賦形剤のいくつかまたは全てをブレンドし、その後、当該粉末を粒子に凝集させる液体、典型的には水の存在下で混合する。粒子を、篩にかけ、且つ/または粉砕して乾燥させ、その後、篩にかけ、且つ/または粉砕して所望の粒径とする。その後粒子を錠剤化/圧縮するか、あるいは錠剤化の前に別の賦形剤、例えば、流動促進剤及び/または滑剤を、加えてよい。
【0066】
錠剤化組成物は、乾式混合によって定法で調製してよい。例えば、活性剤と賦形剤とのブレンド組成物を、スラグまたはシート状にコンパクト化した後、コンパクト化粒子状に粉砕してもよい。コンパクト化粒子は、次いで錠剤状に圧縮してよい。
【0067】
乾式造粒に代えて、ブレンド組成物を、直接圧縮技術を用いて、直接コンパクト化投薬形態に圧縮してもよい。直接圧縮によれば、粒子のない、より均一な錠剤が製造される。直接圧縮錠剤化に特に適した賦形剤には、マイクロクリスタリンセルロース、スプレー乾燥ラクトース、リン酸水素カルシウム二水和物、及びコロイド状シリカが含まれる。これら及び別の賦形剤の、直接圧縮錠剤化における適切な使用は、特に直接圧縮錠剤化の製剤研究の経験及び技術を有する当業者には既知である。
【0068】
本発明のカプセル充填物は、錠剤化に関して記載された前述のあらゆるブレンド及び粒子を含んでよいが、これらには最後の錠剤化工程は行わない。
【0069】
さらにまた、本発明による結晶形態は、哺乳類、好ましくはヒトへの注入による投与のために製剤化することができる。本発明による結晶形態は、例えば、注入のための粘性の液状溶液または懸濁液として製剤化してよい。製剤は、溶媒を含んでよい。こうした溶媒のための検討事項の中には、様々なpHレベルでの該溶媒の物理的及び化学的安定性、粘度(注射針通過性をもたらす)、流動性、沸点、混和性、及び純度が含まれる。適当な溶媒には、アルコールUSP、ベンジルアルコールNF、ベンジルベンゾエートUSP、及び蓖麻子油USPが含まれる。この製剤には、付加的物質、例えば、バッファー、溶解剤、酸化防止剤等をさらに加えてよい。Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery Systems, 7th Ed.
【0070】
本発明はまた、本発明による結晶形態を必要とする哺乳類、好ましくはヒトの治療方法に使用しようとする前記結晶形態を、任意に別の多形相または共結晶と共に含む医薬品製剤を提供する。本発明の医薬品組成物は、結晶形態のTDFA 2:1を含む。本発明による結晶形態は、こうした医薬品組成物の治療のための有効量を、本明細書中に前述される病気に罹患した哺乳類に投与する工程を含む、哺乳類の治療方法において使用してよい。本発明は、さらに、本明細書中に前述される病気、特にHIVの治療のための薬剤の調製のための、本発明の結晶形態の使用に関する。
【0071】
所定の好ましい実施態様に関して本発明を説明したが、別の実施態様は、当業者が本明細書を勘案することによって明らかになるであろう。本発明は、本発明の化合物の調製を詳細に説明する、以下の実施例を参照して更に規定される。当業者には、原材料と方法とのいずれについても、本発明の範囲から逸脱することなく多数の変形を実施しうることが明らかであろう。
【実施例】
【0072】
(実験条件)
X線粉末回折:
XRPDパターンが、Avantium technologies社(オランダ)製のT2ハイスループットXRPD装置を使用して得られた。Hi-Starエリア型検出器を装備した、Bruker GADDS回折計に、プレートを取り付けた。XRPDプラットフォームに、長い格子面間隔についてはベヘン酸銀を用い、短い格子面間隔についてはコランダムを用いて較正した。データ収集を、1.5°乃至41.5°の2θ領域内で、単色CuK(α)線(1.54178Å)を用いて室温で行った。各ウェルの回折パターンを、二つの2θ範囲(第一フレームについては1.5°≦2θ≦21.5°、第二フレームについては19.5°≦2θ≦41.5°)において、各フレームについて120秒の露光時間で収集する。当業者であれば、器具、試料調製、または別の要因における差異により、実験差異が生じうることを理解する。典型的には、XRPDデータは、約0.3度2θ、好ましくは約0.2度、更に好ましくは0.1度、更に一層好ましくは0.05度の変動で収集される。これは、X線ピークが重なっていると考えられる場合に影響を与える。
【0073】
高分解能X線粉末回折:
高分解能粉末パターンを、LynxEye固体検出器を装備した、Brag-Brentano配置のD8 Advanceシステムで収集した。データ収集のために使用された光は、ゲルマニウム結晶によって単色化されたCuK(α1=1.54056Å)であった。パターンを、約2-4°2θから開始して約60-65°2θまでの様々な2θ範囲で、0.04-0.16°2θの範囲のステップで、更なる処理を行わずに収集した。全てのパターンを室温、およそ295Kでとった。
【0074】
単結晶X線回折:
適当な単結晶を選択してガラスファイバーに接着し、これをX線回折ゴニオメーターに載せた。X線回折データを、三つの結晶について、120Kの温度及び室温で、KappaCCDシステム及びFR590 X線発生装置(Bruker Nonius社(オランダ、デルフト)製)により発生させたMoKα光を用いて収集した。単位セルパラメータ及び結晶構造を、ソフトウェアパッケージMaXusを使用して決定し、最適化した。
【0075】
熱分析:
融解特性を、熱流束DSC822e機器(Mettler-Toledo GmbH(スイス)社製)で記録した、DSCサーモグラムから得た。DSC822eを、インジウムの小片(融点=156.6℃;デルタ-H(f)=28.45J/g)を用いて温度及びエンタルピーについて較正した。試料を、標準的な40マイクロリットルのアルミニウムパンに封入し、DSCにおいて25℃乃至300℃に、20℃/分の加熱速度で加熱した。流速50ml/分で無水N2気体を用いて、測定の間、DSC装置をパージした。
【0076】
結晶からの水損失または溶媒による質量損失を、TGA/SDTAによって決定した。試料質量を、TGA/SDTA851e機器(Mettler-Toledo GmbH(スイス)製)で加熱する間に観察することにより、質量対温度の曲線が得られた。TGA/SDTA851eを、インジウム及びアルミニウムを用いて温度について較正した。試料を100マイクロリットルのアルミニウムるつぼに計量して仕込み、密閉した。封にピンで孔を空け、このるつぼを、TGAにおいて、20℃/分の加熱速度で25℃乃至300℃に加熱した。無水N2気体を用いてパージした。DSCに基づく融点決定は、±2.0℃、好ましくは1.0℃の変動性を有する。
【0077】
ラマン分光法:
ラマンスペクトルを、ラマン顕微鏡mW(Kaiser Opticals Inc製)で、0.96cm-1の分解にて、780nmのレーザー及び100の出力を用いて収集した。
【0078】
(実施例)
結晶化実験のための出発物質は、Cipla Ltd.(インド、ムンバイ)製の研究試料として得た。
高分解能X線回折計を使用する、幾つかの市販試料の分析:
テノホビルの市販試料を、地域の薬局(Viread and Truvada)から入手し、錠剤の表面から擦過及び研磨によってコーティングを注意深く除去して、コーティング物質がX線回折パターンに影響しないようにした。Vireadについての二つのXRPDパターンを、別々に調製した試料から、高分解能X線回折計を用いて収集した。第一の試料は、穏やかに粉砕した錠剤から調製し、第二の試料は、コーティング除去及び表面平坦化の後に粉砕されない錠剤から調製した。いずれの試料のXRPDパターンも、第一の試料の粉砕によって誘発される、構造相転移が全く起こっていないことを示した。
【0079】
VireadのX線分析により、これがテノホビルDFをGilead形態1(米国特許第5,935,946号に記載)及びテノホビルジソプロキシルフマレートの共結晶であるTDFA 2:1を含むことが示された。上述の全てのXRPDパターンは、いずれの錠剤においても賦形剤として使用された、ラクトース一水和物の存在を更に示した。表3Aには、粉砕したViread錠剤のXRPDパターンの2θ位置が第一列に、更にGilead形態1(米国特許第5,935,946号)のピーク位置が第二列に、使用された出発物質もしくは実験のピーク位置が第三列に、室温での単結晶構造に基づいて算出されたTDFA 2:1のピーク位置が第四列に、更に、Cambridge Structure Database (REFCODE LACTOS01)に見られる単結晶構造に基づいて算出されたラクトース一水和物のピーク位置が第五列にまとめられている。
【0080】
その粉砕錠剤のXRPDパターンを1つ収集した、Truvada(詳細な表は記載せず)のXRPDパターンを研究したところ、同様の結論が導き出された。このXRPDパターンでは、エムトリシタビンの2θピーク位置もまた観察された。
【0081】
【表3A】
【表3B】
【0082】
マイクロリットルスケールでのTDFA2:1の結晶化
少量の、約2-3mgの市販の出発物質を、プレートウェルに仕込んだ。出発物質は、テトラヒドロフラン/水(80/20 v/v)混合物中に、ストック添加(stock-dose)した。溶媒を、20kPa未満で約45乃至75時間に亘る蒸発により除去し、出発物質を乾燥させた。結晶化溶媒または結晶化溶媒の混合物(50/50 v/v)を、乾燥出発物質を入れたウェルに少量ずつ室温にて加え、40mlの全容積及び50または80mg/mlのストック濃度とした。溶液を加熱し、60℃に30分間に亘って維持した。次いで、約1℃/時間または50℃/時間の冷却速度で、5℃または20℃の最終温度にまで制御冷却を行い、この温度に1、48、75、117、または139時間に亘って維持した。その後、溶媒を20kPaの圧力下、室温にて、48-120時間に亘って蒸発させた。得られた残渣を、X線粉末回折、DSC、及びTG-MSによって分析した。使用した溶媒は、表Iの通りである。HPLCバイアル中での特定の実験では、301.6mgの出発物質を2-メチル-4-ペンタノールに溶解させた。溶液を、60℃に30分間加熱した。次いで、約1℃/時間の冷却速度で室温(約22℃)にまで制御冷却を行い、この温度に48時間に亘って維持した。次いで、固形物質を遠心分離によって上澄み溶液から分離させた。固形物質のXRPD測定により、これがD形態であることが判明した。上澄み溶液を蒸発させ、残渣のXRPDによって、これがフマル酸及び少量のD形態であることが判明した。この実験により、1:1塩であるテノホビルDFから2:1共結晶であるTDFA2:1への転化の際に、過剰のフマル酸が確認される。
【0083】
熱時濾過を用いるミリリットルスケールでのTDFA2:1の結晶化
少量の、約70-75mgの市販の出発物質を、HPLCバイアルに仕込んだ。結晶化溶媒(または溶媒の50/50 v/v混合物)を、少量ずつ、乾燥出発物質を入れたバイアルに室温にて加え、200-1000マイクロリットルの全容積とした。用いた溶媒及び条件は、表IIの通りである。その後、溶液を、60分間について20乃至60℃の速度で加熱し、この温度で濾過した。濾液を1.1または50℃/時間で3または20℃に冷却し、この温度を24時間維持した。次いで、溶媒を、20kPaの圧力下で、20-25℃にて、15-200時間に亘ってバイアルから蒸発させた(表II参照、0.2kPaで500時間に亘る、(R)-(-)-2-オクタノールの場合)。得られた残渣を、X線粉末回折、DSC、及びTGAによって分析した。
【0084】
貧溶媒添加を用いるミリリットルスケールでのTDFA2:1の結晶化
貧溶媒添加実験を、2つの異なるプロトコルに従って行った。各溶媒についての第一プロトコル(正方向貧溶媒添加)により、スラリーを常温で調製し、これを約17-19時間に亘って平衡させた後に濾過してバイアルに入れた。貧溶媒を、1:1の溶媒:貧溶媒比を用いて添加した。この比は、沈降が全く起こらない場合には、引き続き貧溶媒を用いて1:4に増大させた。添加の時間間隔は1時間であった。貧溶媒の総容積は、飽和溶液の容積と同等であった。
【0085】
第二プロトコル(逆方向貧溶媒添加)としては、スラリーを常温で調製し、これを約17-19時間に亘って平衡させた後に濾過して4つのバイアルのセットに入れた。これらの各バイアルの内容物を、貧溶媒を入れたバイアルに加えた。飽和溶液の4つのバイアルの総容積は、貧溶媒の容積と同等であった。添加の時間間隔は1時間であった。
【0086】
沈降物を遠心分離によって回収し、固形生成物を乾燥させてXRPDによって分析した。沈降が全く起こらない場合には、溶液を96-314時間に亘って室温で蒸発させ、残渣をXRPDによって分析した。実験の詳細については、表IIIを参照のこと。
【0087】
スラリー結晶化を用いる、ミリリットルスケールでのTDFA2:1の結晶化
約50mgの出発物質を使用して、溶媒と共にスラリーを作製した(表4参照)。スラリーを、表4に示される通り、2及び10日間の時間間隔で室温または35℃にて撹拌した。この物質を、固形形態の変化を確認するためにXRPDによって検査した。同様のスラリー実験によって得られた物質のXRPDパターンにおいて、フマル酸の強度ピークが、約28.7度2θに観察されたが、これは、1:1テノホビルDFから2:1共結晶への転化の結果としての、スラリー物質中の過剰のフマル酸を示す。
【0088】
【表4】
【0089】
水浴中、室温及び35℃でのスラリー実験により、2日後には出発物質から形態TDFA2:1への添加がもたらされた。10日後のスラリー中の物質のXRPD測定により、固体形態が、依然として、形態TDFA2:1であることが判明した。出発物質を、1,4-ジオキサン及びアセトニトリル中とした、室温でのスラリー実験は、2日及び10日後にいかなる固形形態転化ももたらさなかった。
【0090】
シード結晶化を用いる、ミリリットルスケールでのTDFA2:1の結晶化
3つのタイプのシード実験を、下記の通り行った。
タイプ1
スラリーを、室温にて、約100mgの出発物質を用いて作製した。スラリーを室温で濾過し、少量の約2mgの対応するシードを添加した。溶液を、室温または5℃に、一晩維持した。次いで、溶液を蒸発させ、固形物質をXRPDによって検査した。
タイプ2
実験を、貧溶媒添加の例に記載した通りに、以下の変更と共に行った:沈降の直後に、少量の約2mgの対応するシードを、溶液に添加した。溶液を、室温または5℃に、一晩維持した。次いで、溶液を蒸発させ、固形物質をXRPDによって検査した。
タイプ3
スラリーを、室温にて、約100mgの出発物質を用いて作製した。少量の約5mgの対応するシードを添加した。スラリーを約1時間撹拌した後、これを2日間室温に維持した。次いで、溶液を蒸発させ、固形物質をXRPDによって検査した。
各実験で使用した具体的条件及びシードは、表5に示される。
【0091】
【表5】
【0092】
ミリリットルスケールでのTDFA2:1の結晶化
2,2,2トリフルオロエタノールから:
少量の、約15.8mgの出発物質を、HPLCバイアルに仕込んだ。2,2,2トリフルオロエタノールを、少量ずつ、乾燥出発物質を入れたバイアルに室温にて加え、1000マイクロリットルの全容積とした。バイアルを振盪し、溶解度を目視で評価した。溶液を加熱し、60℃に30分間に亘って維持した。次いで、溶媒を、減圧下で20-25℃にて、バイアルから蒸発させた。蒸発時間及び圧力は、20kPaにて22.5時間であった。蒸発を、4.4kPaにて71時間に亘って継続させた。得られた残渣を、X線粉末回折、DSC、及びTGAによって分析した。
【0093】
アセトンから:
少量の、約15.3mgの出発物質を、HPLCバイアルに仕込んだ。溶媒のアセトンを、少量ずつ、乾燥出発物質を入れたバイアルに室温にて加え、1000マイクロリットルの全容積とした。バイアルを振盪し、溶解度を目視で評価した。次いで、溶媒を、減圧下で20-25℃にて、バイアルから蒸発させた。蒸発時間及び圧力は、20kPaにて22.5時間であった。得られた残渣を、X線粉末回折、DSC、及びTGAによって分析し、TDFA2:1として同定した。
【0094】
ジクロロメタンから:
少量の、約12.4mgの出発物質を、HPLCバイアルに仕込んだ。溶媒のジクロロメタンを、少量ずつ、乾燥出発物質を入れたバイアルに室温にて加え、1000マイクロリットルの全容積とした。バイアルを振盪し、溶解度を目視で評価した。溶液を加熱し、60℃に30分間に亘って維持した。次いで、溶媒を、減圧下で20-25℃にて、バイアルから蒸発させた。蒸発時間及び圧力は、20kPaにて22.5時間であった。得られた残渣を、X線粉末回折、DSC、及びTGAによって分析し、テノホビルDF形態のTDFA2:1として同定した。
【0095】
ニトロメタンから:
少量の、約15.9mgの出発物質を、HPLCバイアルに仕込んだ。溶媒のニトロメタンを、少量ずつ、乾燥出発物質を入れたバイアルに室温にて加え、1000マイクロリットルの全容積とした。バイアルを振盪し、溶解度を目視で評価した。溶液を加熱し、60℃に30分間に亘って維持した。次いで、溶媒を、減圧下で20-25℃にて、バイアルから蒸発させた。蒸発時間及び圧力は、20kPaにて22.5時間であった。得られた残渣を、X線粉末回折、DSC、及びTGAによって分析し、テノホビルDF形態のTDFA2:1として同定した。
【0096】
水から:
少量の、約16.9mgの出発物質を、HPLCバイアルに仕込んだ。溶媒の水を、少量ずつ、乾燥出発物質を入れたバイアルに室温にて加え、1000マイクロリットルの全容積とした。バイアルを振盪し、溶解度を目視で評価した。溶液を加熱し、60℃に30分間に亘って維持した。次いで、溶媒を、減圧下で20-25℃にて、バイアルから蒸発させた。蒸発時間及び圧力は、20kPaにて22.5時間であった。蒸発を、4.4kPaにて71時間に亘って継続させた。得られた残渣を、X線粉末回折、DSC、及びTGAによって分析し、テノホビルDF形態のTDFA2:1として同定した。
【0097】
動的蒸気吸着(DVS)
水分吸着等温線を、Surface Measurement Systems(英国、ロンドン)社製のDVS-1システムを用いて測定した。様々な形態の固形物質の水分取込における相違により、増大する相対湿度についての様々な固形形態の相対安定性における相違が示される。実験は、25℃の一定温度で行った。
【0098】
約11.5mgの形態TDFA2:1の試料を、DVSパン内に広げた。試料を、相対湿度0%にて7時間に亘って乾燥させた。次いで、チャンバーの相対湿度を5%単位毎に0%から95%に増大させ、水蒸気の吸着を観察した。試料を、各段階に1時間に亘って維持した。次いで、相対湿度を5%単位毎に0%にまで低減させ、各段階に1時間に1時間維持することによって、吸着を観察した。吸着−脱離サイクルのグラフは、図3に示される。水蒸気の総取込は約0.8%であったが、これは吸湿性についての業界基準に合致する。購入したままの出発物質を用いる同様のDVS実験では、総蒸気取込は約4%であったが、これは製剤において望ましくなく、更なる対策を要する。実験の終わりに、固形物質をXRPDにより測定したところ、構造には全く変化のないことが判明した(図4)。
【0099】
実施例
TDFA 2:1とULT 1との薬物動態比較研究
TDFA 2:1及びULT 1のバッチを、μM篩いで篩うことによって同等の結晶サイズをもたせて調製した。小型のセルロースカプセルに、およそ15mgのテノホビルDF形態TDFA 2:1とテノホビルULT Yとのいずれかを充填した。それぞれおよそ300グラムの12匹のオスのWisterラットに、形態TDFA 2:1とテノホビルULT 1とのいずれかのカプセル1つを、経口チューブにより投与し、次いで1mlの水道水を投与した。各ラットから、一定の間隔をおいて、尾静脈の穿刺によって少量の血液を採取した。血液試料は、更なる処理のために、即座に液体窒素中で凍結させた。
【0100】
全ての試料を採取した後、各試料の血漿調製物を作製した。血漿試料を、LC-MS-MSによる分析のために、そのテノホビル含量について更に検査した。採取の効率性を、ラット血漿試料に既知量のテノホビルを混ぜることによる比較によって決定した。テノホビル(テノホビルDFの活性代謝物)の濃度を、較正曲線に対してLC-MS-MSを用いて、各試料において定量した。薬物動態の比較結果を、図5に示す。PKデータから、テノホビルジソプロキシルのヘミフマレート共結晶であるTDFA 2:1は、市販のテノホビルジソプロキシルのフマル酸塩であるテノホビルジソプロキシル形態ULT 1と、生物学的に同等であると結論づけられた。
【0101】
表3:テノホビルDF形態TDFA 2:1の最終座標及び等価等方変位
【表6A】
【表6B】
【表6C】
【表6D】
【0102】
【表7】
【0103】
【表8】
【0104】
【表9】
【特許請求の範囲】
【請求項1】
テノホビルジソプロキシルとフマル酸との、フマル酸に対するテノホビルジソプロキシルの割合が約2:1である、組成物(TDFA 2:1)。
【請求項2】
共結晶である、請求項1に記載の組成物。
【請求項3】
共結晶が、120K乃至室温の温度にて共結晶である、請求項1乃至3のいずれか一項に記載の共結晶TDFA 2:1。
【請求項4】
・ 実質的に表1及び/または図1Aに示される通りであるXRPDパターン、
・ 実質的に図1Bに示される通りであるDSC、
・ 実質的に図1Cに示される通りであるTGA、
・ 実質的に図1Eに示される通りである、単結晶構造、
の1つまたは複数によって特徴付けられる、請求項2または3に記載の共結晶TDFA 2:1。
【請求項5】
実質的に純粋形態である、請求項2、3、または4に記載の共結晶TDFA 2:1。
【請求項6】
・ 表Iに記載される適当な溶媒またはその混合物に、テノホビルDFを溶解させるかまたは混入し、溶媒の蒸発により共結晶TDFA 2:1を結晶化させる工程、及び/または
・ 表IIに記載される適当な溶媒またはその混合物に、テノホビルDFを溶解させるかまたは混入し、飽和溶液の冷却及び/または蒸発結晶化により共結晶TDFA 2:1を結晶化させる工程、及び/または
・ 表IIIに記載される適当な溶媒またはその混合物に、テノホビルDFを溶解させるかまたは混入し、表IIIに記載される貧溶媒添加により共結晶TDFA 2:1を結晶化させる工程、及び/または
・ 適当な溶媒またはその混合物にテノホビルDFを溶解させるかまたは混入し、スラリー結晶化及び/またはシード結晶化によりテノホビルDF形態TDFA 2:1を結晶化させる工程、
を含む、共結晶TDFA 2:1の調製方法。
【請求項7】
・ 7.9、9.8、11.0、12.0、13.7、14.3、16.1、16.8、18.0、19.2、20.4、21.2、21.7、22.6、23.4、24.3、25.4、27.6度2θ±0.3度2θ、好ましくは±約0.2度2θ、更に好ましくは±約0.1度2θ、更にいっそう好ましくは±約0.05度2θからなる群より選択される、少なくとも1つ、好ましくは少なくとも2つ、更に好ましくは少なくとも3つ、更にいっそう好ましくは少なくとも4つのX線粉末回折ピーク、
・ DSCにおける117.0±2℃での特徴ピーク、
の1つまたは複数によって特徴付けられる、共結晶TDFA 2:1。
【請求項8】
テノホビルDFを、2,2,2トリフルオロエタノール、アセトン、ジクロロメタン、ニトロメタン、または水に溶解させる工程、及び溶媒の蒸発によって共結晶TDFA 2:1を結晶化させる工程を含む、共結晶TDFA 2:1の調製方法。
【請求項9】
・ 表Iに記載の通り、適当な溶媒またはその混合物にテノホビルジソプロキシルフマレート 1:1を溶解させるかまたは混入し、溶媒の蒸発により共結晶TDFA 2:1を結晶化させる工程、及び/または
・ 表IIに記載の通り、適当な溶媒またはその混合物にテノホビルジソプロキシルフマレート 1:1を溶解させるかまたは混入し、飽和溶液の冷却及び/または蒸発結晶化により共結晶TDFA 2:1を結晶化させる工程、及び/または
・ 表IIIに記載の通り、適当な溶媒またはその混合物にテノホビルジソプロキシルフマレート 1:1を溶解させるかまたは混入し、表IIIに記載の通り貧溶媒添加により共結晶TDFA 2:1を結晶化させる工程、及び/または
・ 適当な溶媒またはその混合物にテノホビルジソプロキシルフマレート 1:1を溶解させるかまたは混入し、スラリー結晶化及び/またはシード結晶化によりテノホビルDF形態TDFA 2:1を結晶化させる工程、
を含む、共結晶TDFA 2:1の調製方法。
【請求項10】
溶媒が、水性溶媒、好ましくは水である、請求項9に記載の方法。
【請求項11】
テノホビルジソプロキシルフマレート1:1を水性溶媒、好ましくは水と接触させる工程を含む、実質的に純粋形態での共結晶TDFA 2:1の調製方法。
【請求項12】
テノホビルジソプロキシルフマレート1:1を実質的に含まない、TDFA 2:1を含む医薬製剤。
【請求項13】
5.5度2θ±0.1度2θにX線ピークを有することによって特徴付けられる固体形態を実質的に含まない、請求項12に記載の医薬製剤。
【請求項14】
共結晶TDFA 2:1の、薬剤としての使用。
【請求項15】
共結晶TDFA 2:1の、HIVの治療のための薬剤の調製における使用。
【請求項16】
共結晶TDFA 2:1の、HIVの治療における使用。
【請求項17】
共結晶TDFA 2:1の、別の医薬品成分、好ましくは抗HIV剤、好ましくはエファビレンツ及び/またはエムトリシタビン及び/またはTMC114との組み合わせでの使用。
【請求項1】
テノホビルジソプロキシルとフマル酸との、フマル酸に対するテノホビルジソプロキシルの割合が約2:1である、組成物(TDFA 2:1)。
【請求項2】
共結晶である、請求項1に記載の組成物。
【請求項3】
共結晶が、120K乃至室温の温度にて共結晶である、請求項1乃至3のいずれか一項に記載の共結晶TDFA 2:1。
【請求項4】
・ 実質的に表1及び/または図1Aに示される通りであるXRPDパターン、
・ 実質的に図1Bに示される通りであるDSC、
・ 実質的に図1Cに示される通りであるTGA、
・ 実質的に図1Eに示される通りである、単結晶構造、
の1つまたは複数によって特徴付けられる、請求項2または3に記載の共結晶TDFA 2:1。
【請求項5】
実質的に純粋形態である、請求項2、3、または4に記載の共結晶TDFA 2:1。
【請求項6】
・ 表Iに記載される適当な溶媒またはその混合物に、テノホビルDFを溶解させるかまたは混入し、溶媒の蒸発により共結晶TDFA 2:1を結晶化させる工程、及び/または
・ 表IIに記載される適当な溶媒またはその混合物に、テノホビルDFを溶解させるかまたは混入し、飽和溶液の冷却及び/または蒸発結晶化により共結晶TDFA 2:1を結晶化させる工程、及び/または
・ 表IIIに記載される適当な溶媒またはその混合物に、テノホビルDFを溶解させるかまたは混入し、表IIIに記載される貧溶媒添加により共結晶TDFA 2:1を結晶化させる工程、及び/または
・ 適当な溶媒またはその混合物にテノホビルDFを溶解させるかまたは混入し、スラリー結晶化及び/またはシード結晶化によりテノホビルDF形態TDFA 2:1を結晶化させる工程、
を含む、共結晶TDFA 2:1の調製方法。
【請求項7】
・ 7.9、9.8、11.0、12.0、13.7、14.3、16.1、16.8、18.0、19.2、20.4、21.2、21.7、22.6、23.4、24.3、25.4、27.6度2θ±0.3度2θ、好ましくは±約0.2度2θ、更に好ましくは±約0.1度2θ、更にいっそう好ましくは±約0.05度2θからなる群より選択される、少なくとも1つ、好ましくは少なくとも2つ、更に好ましくは少なくとも3つ、更にいっそう好ましくは少なくとも4つのX線粉末回折ピーク、
・ DSCにおける117.0±2℃での特徴ピーク、
の1つまたは複数によって特徴付けられる、共結晶TDFA 2:1。
【請求項8】
テノホビルDFを、2,2,2トリフルオロエタノール、アセトン、ジクロロメタン、ニトロメタン、または水に溶解させる工程、及び溶媒の蒸発によって共結晶TDFA 2:1を結晶化させる工程を含む、共結晶TDFA 2:1の調製方法。
【請求項9】
・ 表Iに記載の通り、適当な溶媒またはその混合物にテノホビルジソプロキシルフマレート 1:1を溶解させるかまたは混入し、溶媒の蒸発により共結晶TDFA 2:1を結晶化させる工程、及び/または
・ 表IIに記載の通り、適当な溶媒またはその混合物にテノホビルジソプロキシルフマレート 1:1を溶解させるかまたは混入し、飽和溶液の冷却及び/または蒸発結晶化により共結晶TDFA 2:1を結晶化させる工程、及び/または
・ 表IIIに記載の通り、適当な溶媒またはその混合物にテノホビルジソプロキシルフマレート 1:1を溶解させるかまたは混入し、表IIIに記載の通り貧溶媒添加により共結晶TDFA 2:1を結晶化させる工程、及び/または
・ 適当な溶媒またはその混合物にテノホビルジソプロキシルフマレート 1:1を溶解させるかまたは混入し、スラリー結晶化及び/またはシード結晶化によりテノホビルDF形態TDFA 2:1を結晶化させる工程、
を含む、共結晶TDFA 2:1の調製方法。
【請求項10】
溶媒が、水性溶媒、好ましくは水である、請求項9に記載の方法。
【請求項11】
テノホビルジソプロキシルフマレート1:1を水性溶媒、好ましくは水と接触させる工程を含む、実質的に純粋形態での共結晶TDFA 2:1の調製方法。
【請求項12】
テノホビルジソプロキシルフマレート1:1を実質的に含まない、TDFA 2:1を含む医薬製剤。
【請求項13】
5.5度2θ±0.1度2θにX線ピークを有することによって特徴付けられる固体形態を実質的に含まない、請求項12に記載の医薬製剤。
【請求項14】
共結晶TDFA 2:1の、薬剤としての使用。
【請求項15】
共結晶TDFA 2:1の、HIVの治療のための薬剤の調製における使用。
【請求項16】
共結晶TDFA 2:1の、HIVの治療における使用。
【請求項17】
共結晶TDFA 2:1の、別の医薬品成分、好ましくは抗HIV剤、好ましくはエファビレンツ及び/またはエムトリシタビン及び/またはTMC114との組み合わせでの使用。
【図1A】

【図1B】
【図1C】
【図1D】
【図1E】
【図1F】
【図2A】
【図2B】
【図2C】
【図3】
【図4】
【図5】
【図1B】
【図1C】
【図1D】
【図1E】
【図1F】
【図2A】
【図2B】
【図2C】
【図3】
【図4】
【図5】
【公表番号】特表2010−527996(P2010−527996A)
【公表日】平成22年8月19日(2010.8.19)
【国際特許分類】
【出願番号】特願2010−509283(P2010−509283)
【出願日】平成20年5月21日(2008.5.21)
【国際出願番号】PCT/NL2008/000132
【国際公開番号】WO2008/143500
【国際公開日】平成20年11月27日(2008.11.27)
【出願人】(509322100)ウルティモルフィクス・テクノロジーズ・ベー・フェー (3)
【Fターム(参考)】
【公表日】平成22年8月19日(2010.8.19)
【国際特許分類】
【出願日】平成20年5月21日(2008.5.21)
【国際出願番号】PCT/NL2008/000132
【国際公開番号】WO2008/143500
【国際公開日】平成20年11月27日(2008.11.27)
【出願人】(509322100)ウルティモルフィクス・テクノロジーズ・ベー・フェー (3)
【Fターム(参考)】
[ Back to top ]