
トランスジェニック動物、その作出方法及びそのための核酸
【課題】 外来遺伝子が導入されたか否かを容易かつ明瞭に判別でき、マーカータンパク質が生体の中の極限られた局所にしか発現しない、トランスジェニック動物の作出方法を提供すること。
【解決手段】 トランスジェニック動物の作出方法は、所望の外来遺伝子と、宿主細胞内で該外来遺伝子の発現を制御するプロモーターを含む核酸を受精卵若しくはクローン卵又は胚に導入し、該受精卵若しくはクローン卵又は胚から個体を発生させるトランスジェニック動物の作出方法において、水晶体において特異的に発現する遺伝子のプロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される、可視的に発現の有無を判定できるマーカータンパク質をコードするマーカー遺伝子を、前記核酸に連結して導入することを特徴とする。
【解決手段】 トランスジェニック動物の作出方法は、所望の外来遺伝子と、宿主細胞内で該外来遺伝子の発現を制御するプロモーターを含む核酸を受精卵若しくはクローン卵又は胚に導入し、該受精卵若しくはクローン卵又は胚から個体を発生させるトランスジェニック動物の作出方法において、水晶体において特異的に発現する遺伝子のプロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される、可視的に発現の有無を判定できるマーカータンパク質をコードするマーカー遺伝子を、前記核酸に連結して導入することを特徴とする。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、所望の外来遺伝子が導入されたか否かを簡便に判定することができるトランスジェニック動物、その作出方法及びそのための核酸に関する。
【背景技術】
【0002】
近年の遺伝子操作技術の進歩を背景に,疾患に関与する因子の研究、有用生理活性物質の産生など多くの分野においてトランスジェニック、ノックアウトなどの遺伝子改変を受けた動物が作製されている。外来遺伝子(トランスジーン)を導入する方法も格段に進歩したが、これらの遺伝子改変動物を作製する際に、外来遺伝子が導入されていない動物も多数産まれてくるのが現状である。現在、外来遺伝子の導入された個体と導入されていない個体を判別するために、いくつかの方法が使われている。
【0003】
1つの方法は各個体から組織片を採取してゲノムDNAを抽出し、外来遺伝子特異的なプライマーを使ってPCR法により判別していく方法である。この方法は数の多い調査個体全てに対して行なう場合、非常に多くの手間と時間を要する。特に、トランスジェニック胎仔より培養細胞を確立する場合など、短時間で判別を必要とする場合には何らかの改善が必要となる。
【0004】
別の方法では、目的の遺伝子と共にマーカー遺伝子を組み込んだベクターを使い、目的の遺伝子が導入された個体(胚、細胞など)を選択する。特に、外観で判別が付くマーカー遺伝子の使用が好ましく採用されている。具体的には、チキンβ-アクチンプロモーターによりEGFP(Enhanced Green Fluorescent Protein)を発現させるpCX-EGFPベクター等がその使用例の1つとして挙げられる。pCX-EGFPベクターはチキンβ-アクチンプロモーターを使用していることから、ほぼ全ての細胞でEGFPを発現し、484nmの光により励起して510nmの蛍光を発光するので簡便かつ胚(細胞・組織)の破壊を伴わずに遺伝子導入の成否を判定でき、マーカー遺伝子として広く使用されている。pCX-EGFPベクターが導入された動物としてマウス、ラット、ウサギ、ブタなどの出産報告がある。
【0005】
しかしながら、この方法では、ほぼ全身の細胞でマーカーであるEGFPが発現するため、この蛍光タンパク質による生体への悪影響が懸念される。ウサギ、ブタでは、生まれてきたトランスジェニック動物の生存率が10%以下との報告もある。また、作出されたトランスジェニック動物の遺伝子を維持して次の代に伝える技術として体細胞クローン技術が採用され、今後益々広まっていくと考えられるが、蛍光タンパク質マーカーが発現している体細胞をドナーとして用いると、蛍光タンパク質マーカーによるクローン動物への悪影響が懸念される。さらに、動物は透明ではなく、何らかの色を有しているため、EGPFマーカーの発現量が少ない場合には、外観による識別は必ずしも容易ではない。
【0006】
【特許文献1】特開平8-322427号公報
【非特許文献1】Hogan et al.(1994)、Jean Richa(2001)
【非特許文献2】Lacy et al.1983 Brinster et al.1985
【発明の開示】
【発明が解決しようとする課題】
【0007】
本発明の目的は、外来遺伝子が導入されたか否かを容易かつ明瞭に判別でき、マーカータンパク質が生体の中の極限られた局所にしか発現しない、トランスジェニック動物の作出方法、該方法により作出されたトランスジェニック動物及び該方法に用いられる核酸を提供することである。
【課題を解決するための手段】
【0008】
本願発明者らは、鋭意研究の結果、水晶体において特異的に発現するタンパク質のプロモーターを利用し、このプロモーターの下流に蛍光タンパク質マーカーの遺伝子を配置し、これを目的の外来遺伝子を含む核酸に組み込んで外来遺伝子と共に導入することにより、作出されたトランスジェニック動物の眼に光を当てるだけで、外来遺伝子が導入されたか否かを簡便かつ明瞭に判別することができ、かつ、マーカーは水晶体以外の部分では発現しないことを見出し、本発明を完成した。
【0009】
すなわち、本発明は、所望の外来遺伝子と、宿主細胞内で該外来遺伝子の発現を制御するプロモーターを含む核酸を受精卵若しくはクローン卵又は胚に導入し、該受精卵若しくはクローン卵又は胚から個体を発生させるトランスジェニック動物の作出方法において、水晶体において特異的に発現する遺伝子のプロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される、可視的に発現の有無を判定できるマーカータンパク質をコードするマーカー遺伝子を、前記核酸に連結して導入することを特徴とする、トランスジェニック動物の作出方法を提供する。また、本発明は、上記本発明の方法により作出され、水晶体内において前記マーカー遺伝子を特異的に発現しているトランスジェニック動物又は水晶体内において前記マーカー遺伝子を特異的に発現するその子孫を提供する。さらに、本発明は、水晶体において特異的に発現する遺伝子のプロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される、可視的に発現の有無を判定できるタンパク質をコードするマーカー遺伝子と、所望の外来遺伝子と、該外来遺伝子の上流に位置し、該外来遺伝子の発現を制御するプロモーターとを具備する、トランスジェニック動物作出用核酸を提供する。さらに、本発明は、水晶体において特異的に発現する遺伝子のプロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される、可視的に発現の有無を判定できるタンパク質をコードするマーカー遺伝子と、所望の外来遺伝子を挿入するための制限酵素部位と、該制限酵素部位の上流に位置し、該制限酵素部位に挿入される外来遺伝子の発現を制御するプロモーターとを具備する、上記本発明の核酸を作出するための核酸を提供する。さらに、本発明は、上記本発明の核酸と、宿主細胞中での複製を可能にする複製開始点を少なくとも含む組換えベクターを提供する。
【発明の効果】
【0010】
本発明により、外来遺伝子が導入されたか否かを容易かつ明瞭に判別でき、マーカータンパク質が生体の中の極限られた局所にしか発現しない、トランスジェニック動物の作出方法が初めて提供された。本発明の方法により作出されたトランスジェニック動物では、その眼を観察するだけで外来遺伝子が導入されたか否かを簡便、容易に判定することができる。水晶体は透明であるので、マーカータンパク質の発現量が比較的少ない場合でも、マーカータンパク質が発現しているか否かを明瞭に判定することが可能である。また、マーカータンパク質は、水晶体においてのみ発現するため、マーカータンパク質による全身的な悪影響を避けることができる。また、体細胞クローン作出用のドナーとして用いる場合でも、水晶体以外の部分の体細胞ではマーカータンパク質が発現していないので、マーカータンパク質による、体細胞クローン動物の発生に対する悪影響を避けることができる。
【発明を実施するための最良の形態】
【0011】
上記の通り、本発明のトランスジェニック動物の作出方法は、通常のトランスジェニック動物の作出方法において、水晶体において特異的に発現する遺伝子のプロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される、可視的に発現の有無を判定できるタンパク質をコードするマーカー遺伝子を、前記外来遺伝子に連結して導入することを特徴とする。
【0012】
ここで、水晶体において特異的に発現する遺伝子のプロモーター(以下、便宜的に「眼特異的プロモーター」と呼ぶことがある)としては、水晶体で特異的に発現するαAクリスタリンのプロモーターを例示することができる。これらのうち、αAクリスタリンのプロモーターが好ましい。なお、ここで、「特異的に」とは、水晶体以外の組織では、発現していないことを意味する。発現の有無は、常法であるRT-PCRにより調べることができ、下記実施例にも具体的に記載されている。
【0013】
αAクリスタリンのプロモーター自体はいくつかの種で公知であり、例えば下記実施例で用いた、マウスのαAクリスタリンのプロモーターを含むDNA断片の塩基配列が配列表の配列番号3に示されている。また、αAクリスタリンのcDNAの塩基配列はより多くの種において知られており、ウシ、ヒツジ、ラット、マウス、アフリカツメガエル、ゼブラフィッシュ等で知られている(GenBank Accession No.は、それぞれ、M26142、AY819022、U47922、AJ310308、D88185、AY035778)。cDNAの5'末端領域の塩基配列がわかっていれば、そのプロモーターを含むDNA断片は、常法であるDNA Walkingにより、市販のキット(例えば、BD Biosciences Clontech社製BD GenomeWalker(商品名)等)を用いて容易に調製可能である。すなわち、ゲノムDNAを異なる複数種類の制限酵素で処理し、生じる各制限酵素断片の5'末端に配列公知のアダプターを連結し、目的の遺伝子のcDNAの5'末端領域と、前記アダプター領域に設定したプライマーセットを用いてPCRを行い、増幅された断片をレポーター遺伝子を含むベクターに挿入し、このベクターを宿主に導入してレポーター遺伝子が発現されるDNA断片を選択することにより、目的の遺伝子のプロモーターを含むDNA断片を得ることができる。これは市販のキットの添付文書に従って容易に実施することができる。なお、プロモーターは、作出するトランスジェニック動物と同種の動物由来のプロモーターを用いることが好ましい。なお、プロモーター配列がどこからどこまでかということは特定困難であるが、プロモーターを含む核酸断片は上記方法により容易に得ることができる。後述のように、本発明のトランスジェニック動物作出用核酸を構築するにあたり、プロモーターのみを単離する必要はなく、プロモーターを含む核酸断片を用いることができる。通常、プロモーターは、転写開始点から100塩基程度上流までの断片に含まれているので、この部分を少なくとも含む断片で、下流の構造遺伝子の発現を制御できるプロモーター含有断片を用いることができる。従って、下記実施例では、マウスαAクリスタリンのプロモーターを含有する核酸断片として、配列番号3に示す、マウスαAクリスタリンの-367〜+51(転写開始点を基準)の断片(配列番号3に示す断片は、この領域の5'側にEcoRVの認識配列(gatatc)、3'側にBamHIの認識配列(ggatcc)が付加されている)をαAプロモーター含有断片として用いているが、このように大きなサイズの断片を用いる必要はなく、-111〜0bp程度の位置の断片をαAプロモーター含有断片として用いることが可能である。もっとも、転写が確実に起きるように、プロモーター含有断片に転写開始点及びその下流の短い領域も含めておくことが好ましく、転写開始点を基準として、15bp〜50bp程度下流まで含めておくことが好ましい。従って、3'末端が転写開始点を基準として、+15bp〜+50bp程度、5'末端が-111bp又はこれよりも上流に位置する(5'側は-100bpよりも数百bp程度上流に位置する断片も利用可能である)核酸断片をプロモーター含有断片として好ましく用いることができる。
【0014】
本発明の方法に用いられる、可視的に発現の有無を判定できるタンパク質としては、従来から広く用いられている各種蛍光タンパク質を採用することができる。好ましい例として、GFP(green fluorescent protein)、EGFP(enhanced green fluorescent protein)、EBFP(enhanced blue fluorescent protein, GenBank Accession No. AX766758)、ECFP(enhanced cyan fluorescent protein, GenBank Accession No. DQ00469)、EYFP(enhanced yellow fluorescent protein, GenBank Accession No. AY995145)、AmCyan1 (Anemonia majano cyan fluorescent protein, GenBank Accession No. AF164821)、ZsGreen1 (Zoanthus sp. green fluorescent protein、GenBank Accession No. AF168422)、ZaYellow1(Zoanthus sp. yellow fluorescent protein、GenBank Accession No. AF168423)、DsRed2(Discosoma sp. Red Fluorescent Protein、GenBank Accession No. AF168419)、AsRed2(Anemonia sulcata red fluorescent protein、GenBank Accession No. AX686890)及びHcRed1(Far-red Fluorescent Protein)等を挙げることができる。これらの蛍光タンパク質は、レポータータンパク質として広く用いられており、特にEGFPをコードするcDNAを含むプラスミドベクターは種々市販されている(例えば、Clonetech社製pEGFP-N1(商品名)等)ので、これらの市販のプラスミドを鋳型としたPCRにより容易にそのcDNA断片(例えば配列番号11)を調製することができる。
【0015】
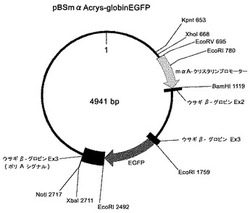
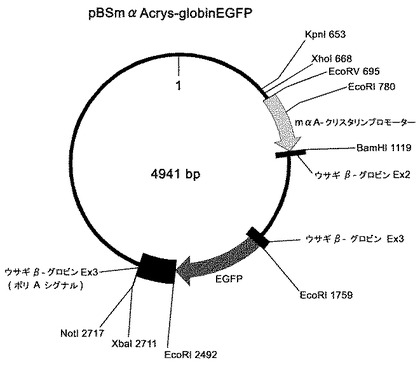
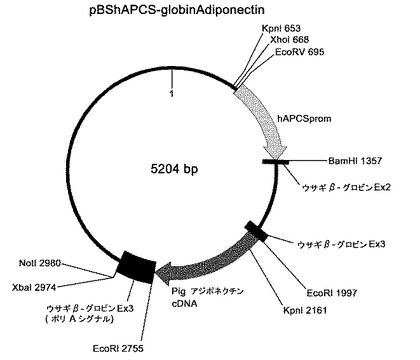
トランスジェニック動物の作出方法として、マイクロインジェクション法が最も広く用いられており、本発明においても、マイクロインジェクション法を好ましく採用することができる。マイクロインジェクション法では、通常、導入すべき目的の外来遺伝子と、その上流に位置し、該外来遺伝子の発現を制御するプロモーターと、該外来遺伝子の下流に位置し、その転写を停止する、polyA配列等のターミネーター配列を含む核酸(以下、このような核酸を便宜的に「外来遺伝子カセット核酸」と呼ぶことがある)を、受精卵の前核に直接注入する。注入する核酸は、環状よりも直鎖状の方が、宿主動物の染色体DNAに挿入され易いことが知られており、通常、直鎖状の核酸が用いられる。例として、肝臓特異的にアジポネクチン(adiponectin)を発現するトランスジェニック動物の作出に用いられている核酸を含む組換えベクターのベクターの遺伝子地図を図2に示す。このベクターでは、市販のベクター(例えばStratagene社のpBluescriptシリーズ等)のマルチクローニング部位に、プロモーター(図2中、hAPCSprom)と、その下流にブタアジポネクチンcDNAと、その下流にウサギβ−グロビン遺伝子のエクソン3中のターミネーター(polyA)が挿入されている。なお、ウサギβ−グロビンのエクソン3を使用しているのは、エクソン3に存在する3'非翻訳領域に、宿主細胞中で転写されたmRNAの安定性を高める(すなわち、mRNAが分解されにくくする)効果があるために採用されているものである。さらに、プロモーターとアジポネクチンcDNAの間には、ウサギβ−グロビンのエクソン2及びエクソン3の一部が位置するが、これは転写開始点と翻訳開始点の間にイントロンが存在するとタンパク発現が高まるためである。ウサギβ−グロビンのエクソン3中にブタアジポネクチンcDNAを挿入すればこのような構造を容易に得ることができる。なお、ウサギβ−グロビン遺伝子由来の領域は、エクソン1を含んでおらず、開始コドンが存在しないため、翻訳はされない。ウサギβ−グロビン遺伝子はその塩基配列も公知であり(GenBank Accession No. V00882)、ウサギのゲノムDNAを鋳型としたPCRにより容易に調製可能である。β−グロビン遺伝子のエクソン2及びエクソン3の他にこのような作用を持つ遺伝子として、α−グロビン遺伝子の3'非翻訳領域、ウシ成長ホルモン(BGH)のpolyAテールも知られており、これらを用いることもできる。図2に示すベクターは環状であるが、マイクロインジェクション法により注入する遺伝子は、直鎖状であることが好ましいので、図2に示すベクターを制限酵素EcoRVとNotIで消化し、生じる2つの断片のうちアジポネクチンcDNAを含有する方の核酸断片を精製して、マイクロインジェクンする。
【0016】
本発明の方法では、このような、トランスジェニック動物に導入すべき外来遺伝子(上記の例ではアジポネクチンcDNA)及びそのプロモーター並びに好ましくは外来遺伝子のターミネーターを含む核酸に、上記した眼特異的プロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される前記マーカー遺伝子と、さらに好ましくはマーカー遺伝子のターミネーターを連結した核酸を導入する。下記実施例において調製したベクターの遺伝子地図を図1に示す。これは上記した図2に示されるベクターと同様な構造を有しており、プロモーターがマウスαAクリスタリンプロモーターに、外来遺伝子がマーカー遺伝子であるEGFPになっているものである。このような構造のベクターを調製した後(具体的な調製方法は下記実施例に詳述)、眼特異的プロモーター、その下流のマーカー遺伝子及びその下流のターミネーターを含む断片(以下、このような核酸断片を便宜的に「マーカーカセット核酸」と呼ぶことがある)をXhoIとNotIで切り出した後にT4 DNA ポリメラーゼ処理により平滑末端にし、図2に示すような、所望の外来遺伝子を含むベクターに挿入することにより(挿入位置は、例えば外来遺伝子プロモーターよりも上流にあるEcoRV部位)、眼特異的プロモーター、マーカー遺伝子、マーカー遺伝子のターミネーター、外来遺伝子のプロモーター、外来遺伝子、外来遺伝子のターミネーターを含む組換えベクターが得られる。挿入部位は、外来遺伝子のターミネーターの下流側であってもよい。あるいは、逆に、図2に示すようなベクターから、外来遺伝子カセット核酸を切り出して、図1に示すベクターに挿入してもよい。このように、外来遺伝子カセット核酸にマーカーカセット核酸を連結した核酸を含むベクターを調製後、外来遺伝子カセット核酸とマーカーカセット核酸を連結した核酸を切り出してマイクロインジェクションする。
【0017】
なお、トランスジェニック動物の作出方法は、マイクロインジェクション法に限定されるものではなく、レトロウイルスベクターやアデノウイルスベクター等の動物用のウイルスベクターに上記核酸を挿入したベクターを受精卵若しくはクローン卵又は胚に導入する公知の方法を採用することもできる。
【0018】
上記したように、眼特異的プロモーターとマーカー遺伝子を含む核酸を、外来遺伝子とそのプロモーター含有核酸に連結した核酸を用いる点を除けば、本発明のトランスジェニック動物の作出方法は、従来のトランスジェニック動物の作出方法と同様に実施することができる。すなわち、上記した核酸断片を、受精卵若しくはクローン卵又は胚に導入する。ここでクローン卵は、除核したレシピエント卵に、体細胞の核(体細胞クローンの場合)又は受精卵の核(受精卵クローンの場合)を移植して得られた卵である。また、胚は、単細胞の卵から、子宮に戻して受胎が可能な胚(好ましくは脱出胚盤胞期胚)までの任意の段階の胚を意味する。もっとも、単細胞の卵の段階で遺伝子導入すれば、トランスジェニック動物の全細胞に遺伝子が含まれるので好ましい。遺伝子を導入した卵又は胚は、好ましくは、常法に従い、桑実期胚まで増殖させた後、動物の子宮に戻し、個体を発生させることができる。
【0019】
本発明の方法により作出された、導入した上記核酸断片が染色体DNA中に挿入されたトランスジェニック動物では、マーカー遺伝子が眼特異的に発現する。マーカータンパク質は、可視的に発現の有無を判定できるものであるので、動物の眼を観察するだけでトランスジェニック動物か否かを容易に判定することができる。例えば、下記実施例のように、マーカー遺伝子としてEGFP遺伝子を用いた場合、波長484nmの光を照射して蛍光が発光されるか否かを調べることにより容易に判定することができる。水晶体は透明であるので、マーカータンパク質の発現量が比較的少ない場合でも、他の組織に比べて明瞭に判定を行なうことができる。マーカー遺伝子と、所望の外来遺伝子は導入した同一の核酸分子中に存在していたのであるから、眼においてマーカー遺伝子の発現が観察された場合には、目的とする外来遺伝子も動物の染色体DNAに挿入されていると考えられる。従って、マーカータンパク質が水晶体において発現しているか否かを調べることにより、外来遺伝子が染色体DNAに挿入された否かを調べることができる。
【0020】
本発明の方法により作出されたトランスジェニック動物では、下記実施例において具体的に記載されるように、水晶体以外の組織では、マーカー遺伝子が発現していない。従って、全身の細胞でマーカー遺伝子が発現して個体に悪影響を与える懸念がない。また、作出されたトランスジェニック動物を核ドナーとして体細胞クローンを作出する場合、ドナーとして用いる細胞ではマーカー遺伝子が発現していないので、マーカー遺伝子が個体発生に対して悪影響を及ぼす懸念も排除される。
【0021】
本発明は、上記した本発明のトランスジェニック動物の作出方法により作出され、水晶体内において前記マーカー遺伝子を特異的に発現しているトランスジェニック動物又は水晶体内において前記マーカー遺伝子を特異的に発現するその子孫をも提供する。ここで、「子孫」とは、通常の有性生殖で得られた子孫のみならず、体細胞クローン技術により、そのトランスジェニック動物と同じ染色体遺伝子を有する体細胞クローン動物をも包含する意味で用いている。なお、体細胞クローン技術は既に常法となっており、ヒツジ、ウシ、ブタ、ヤギ、ウマ、マウス、ネコ等で既に作出されている。本発明の作出方法により作出されたトランスジェニック動物は、染色体DNA中に外来遺伝子とマーカー遺伝子を含むので、これを核ドナーとして用いて得られる体細胞クローン動物は、当然、外来遺伝子とマーカー遺伝子を染色体DNA中に含むものである。
【0022】
本発明は、さらに、上記した、卵又は胚に導入される核酸、すなわち、水晶体において特異的に発現する遺伝子のプロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される、可視的に発現の有無を判定できるタンパク質をコードするマーカー遺伝子と、所望の外来遺伝子と、該外来遺伝子の上流に位置し、該外来遺伝子の発現を制御するプロモーターとを含む、トランスジェニック動物作出用核酸をも提供するものである。
【0023】
さらに、本発明は、このトランスジェニック動物作出用核酸から、外来遺伝子を除去した、このトランスジェニック動物作出用核酸を構築するための核酸をも提供する。このような核酸は、上記したトランスジェニック動物作出用核酸から外来遺伝子を制限酵素で切り出して、再連結したり、上記したトランスジェニック動物作出用核酸の構築方法において、外来遺伝子を含めない方法により容易に構築できる。このような中間体核酸を一旦構築しておけば、この中間体核酸に所望の外来遺伝子を挿入するだけで、所望のトランスジェニック動物作出用核酸を構築することができるので便利である。
【0024】
また、マイクロインジェクション法により注入される核酸断片は、上記のように直鎖状の核酸断片が好ましいが、このような核酸断片を増幅したり、クローニングや精製するために、例えば大腸菌のような宿主細胞中で増殖可能なベクター中に含む組換えベクターを構築すると便利である。本発明は、このような組換えベクターをも提供する。これは、上記した、トランスジェニック動物作出用核酸を構築する方法において、最後にトランスジェニック動物作出用核酸を切り出す前の組換えベクター等であり、上記方法により構築することができる。
【0025】
以下、本発明を実施例に基づきより具体的に説明する。もっとも、本発明は下記実施例に限定されるものではない。
【実施例】
【0026】
1. ベクターの構築
眼の水晶体でマーカーとなるEGFPを発現させるために、図1に示す構造を有するベクター:mαAcrys-globinEGFPを次のようにして構築した。まず、ウサギβ-グロビンのエキソン2〜ポリアデニレーションシグナル部位を含むBamHI-XbaI断片(塩基配列を配列番号6に示す)を調製した。これは、ウサギゲノムDNAを鋳型とし、フォワード側プライマーとしてctgagtgaactgcactgtgac、リバース側プライマーとしてtctagatatgtccttccgagtgagaを用いたPCRにより調製した。なお、リバース側プライマーにはXbaI部位が5'末端に付加してある。PCR ポリメラーゼはPfuTurbo DNA polymerase(商品名、STRATAGENE社)を使用し、「94℃/180秒→(94℃/25秒→72℃/180秒)7サイクル→(94℃/25秒→67℃/180秒)35サイクル→67℃/420秒→4℃/∞」の反応条件で反応を行なった。得られたPCR産物はpCR4Blunt-TOPO(Invitrogen社)にサブクローニングし、その後シークエンスにて配列を確認した。
【0027】
市販のベクターであるpBluescript SK(-)(Stratagene社製)をBamHIとXbaIで処理し、上記で調製したBamHI-XbaI断片を挿入した。そして、ウサギβ-グロビンのエキソン3に存在するEcoRI部位(配列番号6の673nt〜678nt)に、両端にEcoRI認識部位を付加したEGFPのcDNA(733bp)を導入した。なお、両端にEcoRI認識部位を付加したEGFPのcDNAの塩基配列は、配列番号11に示す。このcDNA断片は、次のようにして作製した。EcoRIの認識配列を5'末端に付加したフォワード側プライマーであるEGFP(Eco Kozak-):gaattccgccaccatggtgagcaagとリバース側プライマーであるEGFP(Eco stop-):gaattcttacttgtacagctcgtccを使用し、EGFPのcDNAを含む市販のプラスミドであるpEGFP-N1(クローンテック社)をテンプレートにしてPCRにて調製した。PCRポリメラーゼはPfuTurbo DNA polymerase(商品名、STRATAGENE社)を使用し、「94℃/180秒→(94℃/30秒→50℃/30秒→72℃/60秒)30サイクル→72℃/420秒→4℃/∞」の反応条件で反応を行なった。得られたPCR 産物はpCR4Blunt-TOPO(商品名、Invitrogen社)にサブクローニングし、その後シークエンスにて配列を確認した。最後に、制限酵素EcoRIにより切り出し、EGFPのcDNA(733bp)断片(配列番号11)を作製した。
【0028】
次に、マウスαAクリスタリンのプロモーター(-367〜+51)を含むEcoRV-BamHI断片(塩基配列を配列番号3に示す)を作製した。マウスαAクリスタリンのプロモーターを含有するEcoRV-BamHI断片は、プライマーにそれぞれEcoRVおよびBamHIの認識配列を付加した、フォワード側プライマーmaACrysta-1: gatatcagatctctggaggtttcggag とリバース側プライマーmaACrysta-2: ggatccagggaccgaaggctggcagtgを使用し、マウスゲノムDNAからPCRにて調製した。PCR ポリメラーゼはPfuTurbo DNA polymerase(商品名、STRATAGENE社)を使用し、「94℃/180秒→(94℃/25秒→72℃/180秒)7サイクル→(94℃/25秒→67℃/180秒)35サイクル→67℃/420秒→4℃/∞」の反応条件で反応を行なった。得られたPCR産物はpCR4Blunt-TOPO(商品名、Invitrogen社)にサブクローニングし、その後シークエンスにて配列を確認した。上記のように構築した、pBluescript SK(-)(商品名)を基礎とする組換えベクターをEcoRVとBamHIで消化し、上記EcoRV-BamHI断片を挿入して、図1に示す構造を有する組換えベクターpBSmαAcrys-globinEGFPを得た。
【0029】
2. 供試動物
卵子および精子のドナーとなるB6D2F1マウス、および胚移植レシピエントに用いたICR雌性マウス、偽妊娠誘起交配用精管結紮ICR雄性マウスは全て日本クレア株式会社から購入したものを使用した。これらのマウスは室温22〜24℃、湿度60〜70%、明期と暗期をそれぞれ7:00点灯、19:00消灯となるように設定した飼育環境で飼育した。水、餌はマウスに自由摂取させ、水は水道水を、餌は日本クレア株式会社から購入したCE-2固形飼料(10KGRY放射線滅菌済み)を使用した。なお、購入したマウスは上記の環境に1週間以上馴馳させた後に実験に用いた。B6D2F1マウスは8〜12週齢の個体を用いた。レシピエントに用いたICR雌性マウスは8〜12週齢かつ体重が28〜34gの個体に限定した。
【0030】
3. 前核期卵の採卵
8〜12週令のB6D2F1雌性マウスに7.5IUの妊馬血清性性腺刺激ホルモン(Pregnant Mare's Serum Gonadotropin:PMSG(帝国臓器製薬株式会社))を腹腔内に注射し、47〜48時間後に8.25IUのヒト絨毛性性腺刺激ホルモン(Human Chorionic Gonadotropin:hCG(デンカ製薬株式会社))を同じく腹腔内に注射し、過排卵を誘起した。その後交配用B6D2F1雄性マウスのケージで一晩交配させ、翌朝膣栓(プラグ)が確認できた雌性マウスを前核期卵のドナーマウスとした。hCG注射後20〜21時間でドナーマウスを頚椎脱臼により安楽死させた後、卵管を摘出し、実体顕微鏡下で卵管膨大部の切り裂くことにより卵丘細胞卵子複合体を採取した。卵丘細胞卵子複合体は、ヒアルロニダーゼ(SIGMA CHEMICAL CO.) (CZB培地に25倍濃度で溶解した状態で−20℃保存)処理(750〜1500unit/ml)によって卵丘細胞を取り除いた後、雌雄両前核の形成が確認され、かつ形態学的に正常な卵子のみを選別し、実験の供試卵とした。なお、CZB培地の組成は次の通りである。NaCl 81.62mM、KCl 4.83mM、MgSO4・7H2O 1.18mM、KH2PO4 1.18mM、EDTA・2Na 0.11mM、Na-Lactate 19.76mM、D-Glucose 5.55mM、CaCl2・2H2O 1.7mM、NaHCO3 25.12mM、ピルビン酸ナトリウム 0.27mM、L-グルタミン 1.00mM、BSA 5.00mg/ml、ペニシリン G 75mg/L, ストレプトマイシン 50mg/L
【0031】
4. トランスジェニック動物作出用核酸の調製
上記1で構築した、図1に示す構造を有する組換えベクターpBSmαAcrys-globinEGFPをKpnIとNotIで消化して2つに切断し、消化物をアガロースゲル電気泳動にかけて2つの断片を分離した。遺伝子導入に用いるKpnI(653)からNotI(2717)の断片(図1参照)をアガロースゲルから切り出し、GENECLEAN(商品名)にて精製して、トランスジェニック動物作出用核酸を得た。これを、pH7.5に調整したTE bufferで10ng/μl濃度に希釈して、後述する前核へのマイクロインジェクションに用いた。
【0032】
5. 前核注入法
前核注入法については非特許文献1の方法を参考にした。前核注入法によって染色体上に組み込まれた外来DNAは、1〜数100コピーの長さで1〜複数の遺伝子座のランダムな位置に整列した状態で組み込まれるという報告がなされている。(非特許文献2) そのため注入に用いるプラスミドDNAは環状のままではなく、制限酵素を用いて直鎖状にしたものを用いたほうがその導入効率は向上し、現在でも制限酵素によるプラスミドDNAの切断処理は広く支持されている。前核注入はM2培地 (SIGMA社製、組成:CaCl2・2H2O 0.251g/L、MgSO4 0.165g/L、KCl 0.356g/L、KH2PO4 0.162g/L、NaCl 5.532g/L、NaHCO3 0.35g/L、アルブミン(Bovine FractionV) 4.0g/L、D-グルコース 1.0g/L、HEPES 5.43g/L、フェノールレッドNa塩 0.01g/L、ピルビン酸ナトリウム 0.036g/L、乳酸ナトリウム 4.35g/Lにペニシリン G 60mg/L、ストレプトマイシン 50mg/Lを添加)を用いて行った。前核注入法は、より具体的には次のようにして行なった。
【0033】
ホールディングピペットはガラスキャピラリー(CLARK ELECTROMEDICAL INSTRUMENT GC100-15)をアルコールランプで熱して引き、外径が約100μmの位置をルビーカッターにより切断した。さらに、ガラスキャピラリーをマイクロフォージ(NARISHIGE CO.JAPAN MF-79)のフィラメント上のガラス玉に垂直になるようにセットし、フィラメントを熱して内径が約10μmとなるようにガラスキャピラリー断面を丸めた。作製したホールディングピペットは乾熱滅菌し、使用時まで滅菌保存した。また、インジェクションピペットは乾熱滅菌しておいたガラスキャピラリー(CLARK ELECTROMEDICAL INSTRUMENT GC100TF-15)をキャピラリープラー(SUTTER INSTRUMENT Co. P-97/IVF)を用いて作製した。
【0034】
倒立顕微鏡ステージ上のホットプレートを37.5℃に設定し、ミネラルオイル中に5μlのM2培地ドロップが6〜8個になるように作製したマニピュレーション用ディッシュをステージに乗せた。ホールディングピペットをマイクロインジェクター(NARISHIGE CO.JAPAN IM9B)に、DNA溶液を充填したインジェクションピペットをインジェクター(NARISHIGE CO.JAPAN IM−200)にセットし、両ピペットの先端がM2培地ドロップ内に位置するようにセットした。本発明では、DNA溶液としてKpnI/NotI処理により直鎖状にしたmαAcrys-globinEGFPをpH7.5に調整したTE bufferで10 ng/μl濃度に希釈し、インジェクションピペットに充填した。
【0035】
マニピュレーション用ディッシュ中のM2培地ドロップ内へ20〜30個の受精卵を入れ、インジェクションピペットを受精卵の雄性前核に刺し、上記4で調製した、mαAcrys-globinEGFP由来のトランスジェニック動物作出用核酸溶液を約2pl注入した。M2培地ドロップ内の受精卵へ注入を終えたらCZB培地に受精卵を移し、20〜30分間インキュベータ内で培養して生存判定後、生存卵のみを桑実期胚まで培養した。培養は37.5℃、5%CO2 in air、湿度100%の条件に設定された炭酸ガス培養装置内で行なった。
【0036】
6. レシピエントマウスへの胚移植
桑実期胚まで発生した胚を胚移植に用いた。レシピエントマウスには8〜12週令のICR雌性を用いた。陰部の観察によって発情前期であると確認できたICR雌性マウスを、精管結紮処置を施したICR雄性マウスのケージで一晩交配させ、翌朝プラグが確認できた偽妊娠マウスをレシピエントとして用いた。プラグが確認できた時点を交配0.5日後(0.5 days post coitus :0.5 d.p.c.)とし、桑実期胚は3 d.p.c.の偽妊娠ICR雌性マウス子宮へ移植した。その後19〜20d.p.c.でレシピエントマウスを剖検し、胎仔を回収した。
【0037】
7. トランスジェニックマウスの判定
19〜20d.p.c.で回収した胎仔を解剖して眼を取り出し、実体蛍光顕微鏡にて視軸方向に沿って角膜より水晶体を観察した。観察した全32匹の胎仔のうち、7匹の胎仔の水晶体が蛍光を発していた。また、他の組織についても同様に観察したところ、蛍光を発している組織は観察されなかった。これらのマウスよりゲノムDNAを分離し、PCRにてマーカー遺伝子の確認を行なったところ、EGFP発現ベクター:mαAcrys-globinEGFPが導入されたトランスジェニックマウスと蛍光を発光していたマウスが一致した。
【0038】
8. 組織特異的発現の確認
トランスジェニックマウスと確認されたマウスについて、眼、内臓塊おけるEGFPの発現をRT-PCRで確認した。これは具体的に次のようにして行なった。仮親マウスから摘出したマウス胎児から眼球を摘出して眼組織とし、胸腔及び腹腔内に存在する臓器をまとめて内臓塊として処理した。それぞれの組織にISOGEN(商品名、ニッポンジーン社製)を加えてホモジナイズし、室温にて5分放置後、クロロホルムを加えて12000G、4℃、15分間遠心分離した。二層に分かれた水層(上層)を別のチューブに移し、イソプロピルアルコールを加えてイソプロピルアルコール沈殿を行い、沈殿としてtotal RNAを回収した。Total RNAはDEPC処理水に溶解し、100ng/μlの濃度に調製した。PCRチューブに総RNA 100ng、oligo(dT12-18) 0.5μg、10mM dNTP mix 1μlを加え、DEPC処理水により全量を12μlとした。70℃にて10分間インキュベーションし、すばやく氷上に移した。さらに5X first strand buffer 4μl(Invitrogen社)、0.1M DTT 2μl、RNaseOUT 1μl(商品名、Invitrogen社)を加え、全量を19μlとした。42℃、1分間のプレインキュベーション後、スーパースクリプトIIRNaseH-逆転写酵素(Invitrogen社)を1μl加え、42℃ 50分間インキュベーションした。最後に70℃ 15分間インキュベーションして反応を止め、Ribonuclease H(タカラバイオ社)を加えて第1鎖cDNAを完成させた。
【0039】
得られた第1鎖cDNAをテンプレートとし、フォワード側プライマーにRTRab-b-GB(Ex.2):ggatcctgagaacttcagg、リバース側プライマーにRTEGFP:agtcgtgctgcttcatgtgを使用してPCRを行なった。PCR ポリメラーゼはTaKaRa Ex Taq(タカラバイオ社)を使用し、「94℃/180秒→(94℃/25秒→72℃/180秒)7サイクル→(94℃/25秒→67℃/180秒)32サイクル→72℃/420秒→4℃/∞」の反応条件で反応を行なった。ゲノムに取り込まれたmαAcrys-globinEGFPがテンプレートになった場合は903bpのPCRプロダクトが得られる。また、mαAcrys-globinEGFPから転写されたmRNAはスプライシングを受けるので、mRNA由来のcDNAがテンプレートになった場合は330bpのPCRプロダクトが確認される。今回のRT-PCRの結果、トランスジェニックマウスの眼球由来mRNAから330bpのPCRプロダクトが得られたので、眼球特異的にEGFP mRNAの転写が行なわれていることが確認された。この結果、EGFP遺伝子は眼特異的に働いていることが確認され、それ以外の組織における発現は確認されなかった。
【図面の簡単な説明】
【0040】
【図1】実施例において作製した、マウスαAプロモーターと、EGFP遺伝子を含む組換えベクターの遺伝子地図である。
【図2】外来遺伝子としてアジポネクチン遺伝子を導入するために用いられる組換えベクターの遺伝子地図である。
【技術分野】
【0001】
本発明は、所望の外来遺伝子が導入されたか否かを簡便に判定することができるトランスジェニック動物、その作出方法及びそのための核酸に関する。
【背景技術】
【0002】
近年の遺伝子操作技術の進歩を背景に,疾患に関与する因子の研究、有用生理活性物質の産生など多くの分野においてトランスジェニック、ノックアウトなどの遺伝子改変を受けた動物が作製されている。外来遺伝子(トランスジーン)を導入する方法も格段に進歩したが、これらの遺伝子改変動物を作製する際に、外来遺伝子が導入されていない動物も多数産まれてくるのが現状である。現在、外来遺伝子の導入された個体と導入されていない個体を判別するために、いくつかの方法が使われている。
【0003】
1つの方法は各個体から組織片を採取してゲノムDNAを抽出し、外来遺伝子特異的なプライマーを使ってPCR法により判別していく方法である。この方法は数の多い調査個体全てに対して行なう場合、非常に多くの手間と時間を要する。特に、トランスジェニック胎仔より培養細胞を確立する場合など、短時間で判別を必要とする場合には何らかの改善が必要となる。
【0004】
別の方法では、目的の遺伝子と共にマーカー遺伝子を組み込んだベクターを使い、目的の遺伝子が導入された個体(胚、細胞など)を選択する。特に、外観で判別が付くマーカー遺伝子の使用が好ましく採用されている。具体的には、チキンβ-アクチンプロモーターによりEGFP(Enhanced Green Fluorescent Protein)を発現させるpCX-EGFPベクター等がその使用例の1つとして挙げられる。pCX-EGFPベクターはチキンβ-アクチンプロモーターを使用していることから、ほぼ全ての細胞でEGFPを発現し、484nmの光により励起して510nmの蛍光を発光するので簡便かつ胚(細胞・組織)の破壊を伴わずに遺伝子導入の成否を判定でき、マーカー遺伝子として広く使用されている。pCX-EGFPベクターが導入された動物としてマウス、ラット、ウサギ、ブタなどの出産報告がある。
【0005】
しかしながら、この方法では、ほぼ全身の細胞でマーカーであるEGFPが発現するため、この蛍光タンパク質による生体への悪影響が懸念される。ウサギ、ブタでは、生まれてきたトランスジェニック動物の生存率が10%以下との報告もある。また、作出されたトランスジェニック動物の遺伝子を維持して次の代に伝える技術として体細胞クローン技術が採用され、今後益々広まっていくと考えられるが、蛍光タンパク質マーカーが発現している体細胞をドナーとして用いると、蛍光タンパク質マーカーによるクローン動物への悪影響が懸念される。さらに、動物は透明ではなく、何らかの色を有しているため、EGPFマーカーの発現量が少ない場合には、外観による識別は必ずしも容易ではない。
【0006】
【特許文献1】特開平8-322427号公報
【非特許文献1】Hogan et al.(1994)、Jean Richa(2001)
【非特許文献2】Lacy et al.1983 Brinster et al.1985
【発明の開示】
【発明が解決しようとする課題】
【0007】
本発明の目的は、外来遺伝子が導入されたか否かを容易かつ明瞭に判別でき、マーカータンパク質が生体の中の極限られた局所にしか発現しない、トランスジェニック動物の作出方法、該方法により作出されたトランスジェニック動物及び該方法に用いられる核酸を提供することである。
【課題を解決するための手段】
【0008】
本願発明者らは、鋭意研究の結果、水晶体において特異的に発現するタンパク質のプロモーターを利用し、このプロモーターの下流に蛍光タンパク質マーカーの遺伝子を配置し、これを目的の外来遺伝子を含む核酸に組み込んで外来遺伝子と共に導入することにより、作出されたトランスジェニック動物の眼に光を当てるだけで、外来遺伝子が導入されたか否かを簡便かつ明瞭に判別することができ、かつ、マーカーは水晶体以外の部分では発現しないことを見出し、本発明を完成した。
【0009】
すなわち、本発明は、所望の外来遺伝子と、宿主細胞内で該外来遺伝子の発現を制御するプロモーターを含む核酸を受精卵若しくはクローン卵又は胚に導入し、該受精卵若しくはクローン卵又は胚から個体を発生させるトランスジェニック動物の作出方法において、水晶体において特異的に発現する遺伝子のプロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される、可視的に発現の有無を判定できるマーカータンパク質をコードするマーカー遺伝子を、前記核酸に連結して導入することを特徴とする、トランスジェニック動物の作出方法を提供する。また、本発明は、上記本発明の方法により作出され、水晶体内において前記マーカー遺伝子を特異的に発現しているトランスジェニック動物又は水晶体内において前記マーカー遺伝子を特異的に発現するその子孫を提供する。さらに、本発明は、水晶体において特異的に発現する遺伝子のプロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される、可視的に発現の有無を判定できるタンパク質をコードするマーカー遺伝子と、所望の外来遺伝子と、該外来遺伝子の上流に位置し、該外来遺伝子の発現を制御するプロモーターとを具備する、トランスジェニック動物作出用核酸を提供する。さらに、本発明は、水晶体において特異的に発現する遺伝子のプロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される、可視的に発現の有無を判定できるタンパク質をコードするマーカー遺伝子と、所望の外来遺伝子を挿入するための制限酵素部位と、該制限酵素部位の上流に位置し、該制限酵素部位に挿入される外来遺伝子の発現を制御するプロモーターとを具備する、上記本発明の核酸を作出するための核酸を提供する。さらに、本発明は、上記本発明の核酸と、宿主細胞中での複製を可能にする複製開始点を少なくとも含む組換えベクターを提供する。
【発明の効果】
【0010】
本発明により、外来遺伝子が導入されたか否かを容易かつ明瞭に判別でき、マーカータンパク質が生体の中の極限られた局所にしか発現しない、トランスジェニック動物の作出方法が初めて提供された。本発明の方法により作出されたトランスジェニック動物では、その眼を観察するだけで外来遺伝子が導入されたか否かを簡便、容易に判定することができる。水晶体は透明であるので、マーカータンパク質の発現量が比較的少ない場合でも、マーカータンパク質が発現しているか否かを明瞭に判定することが可能である。また、マーカータンパク質は、水晶体においてのみ発現するため、マーカータンパク質による全身的な悪影響を避けることができる。また、体細胞クローン作出用のドナーとして用いる場合でも、水晶体以外の部分の体細胞ではマーカータンパク質が発現していないので、マーカータンパク質による、体細胞クローン動物の発生に対する悪影響を避けることができる。
【発明を実施するための最良の形態】
【0011】
上記の通り、本発明のトランスジェニック動物の作出方法は、通常のトランスジェニック動物の作出方法において、水晶体において特異的に発現する遺伝子のプロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される、可視的に発現の有無を判定できるタンパク質をコードするマーカー遺伝子を、前記外来遺伝子に連結して導入することを特徴とする。
【0012】
ここで、水晶体において特異的に発現する遺伝子のプロモーター(以下、便宜的に「眼特異的プロモーター」と呼ぶことがある)としては、水晶体で特異的に発現するαAクリスタリンのプロモーターを例示することができる。これらのうち、αAクリスタリンのプロモーターが好ましい。なお、ここで、「特異的に」とは、水晶体以外の組織では、発現していないことを意味する。発現の有無は、常法であるRT-PCRにより調べることができ、下記実施例にも具体的に記載されている。
【0013】
αAクリスタリンのプロモーター自体はいくつかの種で公知であり、例えば下記実施例で用いた、マウスのαAクリスタリンのプロモーターを含むDNA断片の塩基配列が配列表の配列番号3に示されている。また、αAクリスタリンのcDNAの塩基配列はより多くの種において知られており、ウシ、ヒツジ、ラット、マウス、アフリカツメガエル、ゼブラフィッシュ等で知られている(GenBank Accession No.は、それぞれ、M26142、AY819022、U47922、AJ310308、D88185、AY035778)。cDNAの5'末端領域の塩基配列がわかっていれば、そのプロモーターを含むDNA断片は、常法であるDNA Walkingにより、市販のキット(例えば、BD Biosciences Clontech社製BD GenomeWalker(商品名)等)を用いて容易に調製可能である。すなわち、ゲノムDNAを異なる複数種類の制限酵素で処理し、生じる各制限酵素断片の5'末端に配列公知のアダプターを連結し、目的の遺伝子のcDNAの5'末端領域と、前記アダプター領域に設定したプライマーセットを用いてPCRを行い、増幅された断片をレポーター遺伝子を含むベクターに挿入し、このベクターを宿主に導入してレポーター遺伝子が発現されるDNA断片を選択することにより、目的の遺伝子のプロモーターを含むDNA断片を得ることができる。これは市販のキットの添付文書に従って容易に実施することができる。なお、プロモーターは、作出するトランスジェニック動物と同種の動物由来のプロモーターを用いることが好ましい。なお、プロモーター配列がどこからどこまでかということは特定困難であるが、プロモーターを含む核酸断片は上記方法により容易に得ることができる。後述のように、本発明のトランスジェニック動物作出用核酸を構築するにあたり、プロモーターのみを単離する必要はなく、プロモーターを含む核酸断片を用いることができる。通常、プロモーターは、転写開始点から100塩基程度上流までの断片に含まれているので、この部分を少なくとも含む断片で、下流の構造遺伝子の発現を制御できるプロモーター含有断片を用いることができる。従って、下記実施例では、マウスαAクリスタリンのプロモーターを含有する核酸断片として、配列番号3に示す、マウスαAクリスタリンの-367〜+51(転写開始点を基準)の断片(配列番号3に示す断片は、この領域の5'側にEcoRVの認識配列(gatatc)、3'側にBamHIの認識配列(ggatcc)が付加されている)をαAプロモーター含有断片として用いているが、このように大きなサイズの断片を用いる必要はなく、-111〜0bp程度の位置の断片をαAプロモーター含有断片として用いることが可能である。もっとも、転写が確実に起きるように、プロモーター含有断片に転写開始点及びその下流の短い領域も含めておくことが好ましく、転写開始点を基準として、15bp〜50bp程度下流まで含めておくことが好ましい。従って、3'末端が転写開始点を基準として、+15bp〜+50bp程度、5'末端が-111bp又はこれよりも上流に位置する(5'側は-100bpよりも数百bp程度上流に位置する断片も利用可能である)核酸断片をプロモーター含有断片として好ましく用いることができる。
【0014】
本発明の方法に用いられる、可視的に発現の有無を判定できるタンパク質としては、従来から広く用いられている各種蛍光タンパク質を採用することができる。好ましい例として、GFP(green fluorescent protein)、EGFP(enhanced green fluorescent protein)、EBFP(enhanced blue fluorescent protein, GenBank Accession No. AX766758)、ECFP(enhanced cyan fluorescent protein, GenBank Accession No. DQ00469)、EYFP(enhanced yellow fluorescent protein, GenBank Accession No. AY995145)、AmCyan1 (Anemonia majano cyan fluorescent protein, GenBank Accession No. AF164821)、ZsGreen1 (Zoanthus sp. green fluorescent protein、GenBank Accession No. AF168422)、ZaYellow1(Zoanthus sp. yellow fluorescent protein、GenBank Accession No. AF168423)、DsRed2(Discosoma sp. Red Fluorescent Protein、GenBank Accession No. AF168419)、AsRed2(Anemonia sulcata red fluorescent protein、GenBank Accession No. AX686890)及びHcRed1(Far-red Fluorescent Protein)等を挙げることができる。これらの蛍光タンパク質は、レポータータンパク質として広く用いられており、特にEGFPをコードするcDNAを含むプラスミドベクターは種々市販されている(例えば、Clonetech社製pEGFP-N1(商品名)等)ので、これらの市販のプラスミドを鋳型としたPCRにより容易にそのcDNA断片(例えば配列番号11)を調製することができる。
【0015】
トランスジェニック動物の作出方法として、マイクロインジェクション法が最も広く用いられており、本発明においても、マイクロインジェクション法を好ましく採用することができる。マイクロインジェクション法では、通常、導入すべき目的の外来遺伝子と、その上流に位置し、該外来遺伝子の発現を制御するプロモーターと、該外来遺伝子の下流に位置し、その転写を停止する、polyA配列等のターミネーター配列を含む核酸(以下、このような核酸を便宜的に「外来遺伝子カセット核酸」と呼ぶことがある)を、受精卵の前核に直接注入する。注入する核酸は、環状よりも直鎖状の方が、宿主動物の染色体DNAに挿入され易いことが知られており、通常、直鎖状の核酸が用いられる。例として、肝臓特異的にアジポネクチン(adiponectin)を発現するトランスジェニック動物の作出に用いられている核酸を含む組換えベクターのベクターの遺伝子地図を図2に示す。このベクターでは、市販のベクター(例えばStratagene社のpBluescriptシリーズ等)のマルチクローニング部位に、プロモーター(図2中、hAPCSprom)と、その下流にブタアジポネクチンcDNAと、その下流にウサギβ−グロビン遺伝子のエクソン3中のターミネーター(polyA)が挿入されている。なお、ウサギβ−グロビンのエクソン3を使用しているのは、エクソン3に存在する3'非翻訳領域に、宿主細胞中で転写されたmRNAの安定性を高める(すなわち、mRNAが分解されにくくする)効果があるために採用されているものである。さらに、プロモーターとアジポネクチンcDNAの間には、ウサギβ−グロビンのエクソン2及びエクソン3の一部が位置するが、これは転写開始点と翻訳開始点の間にイントロンが存在するとタンパク発現が高まるためである。ウサギβ−グロビンのエクソン3中にブタアジポネクチンcDNAを挿入すればこのような構造を容易に得ることができる。なお、ウサギβ−グロビン遺伝子由来の領域は、エクソン1を含んでおらず、開始コドンが存在しないため、翻訳はされない。ウサギβ−グロビン遺伝子はその塩基配列も公知であり(GenBank Accession No. V00882)、ウサギのゲノムDNAを鋳型としたPCRにより容易に調製可能である。β−グロビン遺伝子のエクソン2及びエクソン3の他にこのような作用を持つ遺伝子として、α−グロビン遺伝子の3'非翻訳領域、ウシ成長ホルモン(BGH)のpolyAテールも知られており、これらを用いることもできる。図2に示すベクターは環状であるが、マイクロインジェクション法により注入する遺伝子は、直鎖状であることが好ましいので、図2に示すベクターを制限酵素EcoRVとNotIで消化し、生じる2つの断片のうちアジポネクチンcDNAを含有する方の核酸断片を精製して、マイクロインジェクンする。
【0016】
本発明の方法では、このような、トランスジェニック動物に導入すべき外来遺伝子(上記の例ではアジポネクチンcDNA)及びそのプロモーター並びに好ましくは外来遺伝子のターミネーターを含む核酸に、上記した眼特異的プロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される前記マーカー遺伝子と、さらに好ましくはマーカー遺伝子のターミネーターを連結した核酸を導入する。下記実施例において調製したベクターの遺伝子地図を図1に示す。これは上記した図2に示されるベクターと同様な構造を有しており、プロモーターがマウスαAクリスタリンプロモーターに、外来遺伝子がマーカー遺伝子であるEGFPになっているものである。このような構造のベクターを調製した後(具体的な調製方法は下記実施例に詳述)、眼特異的プロモーター、その下流のマーカー遺伝子及びその下流のターミネーターを含む断片(以下、このような核酸断片を便宜的に「マーカーカセット核酸」と呼ぶことがある)をXhoIとNotIで切り出した後にT4 DNA ポリメラーゼ処理により平滑末端にし、図2に示すような、所望の外来遺伝子を含むベクターに挿入することにより(挿入位置は、例えば外来遺伝子プロモーターよりも上流にあるEcoRV部位)、眼特異的プロモーター、マーカー遺伝子、マーカー遺伝子のターミネーター、外来遺伝子のプロモーター、外来遺伝子、外来遺伝子のターミネーターを含む組換えベクターが得られる。挿入部位は、外来遺伝子のターミネーターの下流側であってもよい。あるいは、逆に、図2に示すようなベクターから、外来遺伝子カセット核酸を切り出して、図1に示すベクターに挿入してもよい。このように、外来遺伝子カセット核酸にマーカーカセット核酸を連結した核酸を含むベクターを調製後、外来遺伝子カセット核酸とマーカーカセット核酸を連結した核酸を切り出してマイクロインジェクションする。
【0017】
なお、トランスジェニック動物の作出方法は、マイクロインジェクション法に限定されるものではなく、レトロウイルスベクターやアデノウイルスベクター等の動物用のウイルスベクターに上記核酸を挿入したベクターを受精卵若しくはクローン卵又は胚に導入する公知の方法を採用することもできる。
【0018】
上記したように、眼特異的プロモーターとマーカー遺伝子を含む核酸を、外来遺伝子とそのプロモーター含有核酸に連結した核酸を用いる点を除けば、本発明のトランスジェニック動物の作出方法は、従来のトランスジェニック動物の作出方法と同様に実施することができる。すなわち、上記した核酸断片を、受精卵若しくはクローン卵又は胚に導入する。ここでクローン卵は、除核したレシピエント卵に、体細胞の核(体細胞クローンの場合)又は受精卵の核(受精卵クローンの場合)を移植して得られた卵である。また、胚は、単細胞の卵から、子宮に戻して受胎が可能な胚(好ましくは脱出胚盤胞期胚)までの任意の段階の胚を意味する。もっとも、単細胞の卵の段階で遺伝子導入すれば、トランスジェニック動物の全細胞に遺伝子が含まれるので好ましい。遺伝子を導入した卵又は胚は、好ましくは、常法に従い、桑実期胚まで増殖させた後、動物の子宮に戻し、個体を発生させることができる。
【0019】
本発明の方法により作出された、導入した上記核酸断片が染色体DNA中に挿入されたトランスジェニック動物では、マーカー遺伝子が眼特異的に発現する。マーカータンパク質は、可視的に発現の有無を判定できるものであるので、動物の眼を観察するだけでトランスジェニック動物か否かを容易に判定することができる。例えば、下記実施例のように、マーカー遺伝子としてEGFP遺伝子を用いた場合、波長484nmの光を照射して蛍光が発光されるか否かを調べることにより容易に判定することができる。水晶体は透明であるので、マーカータンパク質の発現量が比較的少ない場合でも、他の組織に比べて明瞭に判定を行なうことができる。マーカー遺伝子と、所望の外来遺伝子は導入した同一の核酸分子中に存在していたのであるから、眼においてマーカー遺伝子の発現が観察された場合には、目的とする外来遺伝子も動物の染色体DNAに挿入されていると考えられる。従って、マーカータンパク質が水晶体において発現しているか否かを調べることにより、外来遺伝子が染色体DNAに挿入された否かを調べることができる。
【0020】
本発明の方法により作出されたトランスジェニック動物では、下記実施例において具体的に記載されるように、水晶体以外の組織では、マーカー遺伝子が発現していない。従って、全身の細胞でマーカー遺伝子が発現して個体に悪影響を与える懸念がない。また、作出されたトランスジェニック動物を核ドナーとして体細胞クローンを作出する場合、ドナーとして用いる細胞ではマーカー遺伝子が発現していないので、マーカー遺伝子が個体発生に対して悪影響を及ぼす懸念も排除される。
【0021】
本発明は、上記した本発明のトランスジェニック動物の作出方法により作出され、水晶体内において前記マーカー遺伝子を特異的に発現しているトランスジェニック動物又は水晶体内において前記マーカー遺伝子を特異的に発現するその子孫をも提供する。ここで、「子孫」とは、通常の有性生殖で得られた子孫のみならず、体細胞クローン技術により、そのトランスジェニック動物と同じ染色体遺伝子を有する体細胞クローン動物をも包含する意味で用いている。なお、体細胞クローン技術は既に常法となっており、ヒツジ、ウシ、ブタ、ヤギ、ウマ、マウス、ネコ等で既に作出されている。本発明の作出方法により作出されたトランスジェニック動物は、染色体DNA中に外来遺伝子とマーカー遺伝子を含むので、これを核ドナーとして用いて得られる体細胞クローン動物は、当然、外来遺伝子とマーカー遺伝子を染色体DNA中に含むものである。
【0022】
本発明は、さらに、上記した、卵又は胚に導入される核酸、すなわち、水晶体において特異的に発現する遺伝子のプロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される、可視的に発現の有無を判定できるタンパク質をコードするマーカー遺伝子と、所望の外来遺伝子と、該外来遺伝子の上流に位置し、該外来遺伝子の発現を制御するプロモーターとを含む、トランスジェニック動物作出用核酸をも提供するものである。
【0023】
さらに、本発明は、このトランスジェニック動物作出用核酸から、外来遺伝子を除去した、このトランスジェニック動物作出用核酸を構築するための核酸をも提供する。このような核酸は、上記したトランスジェニック動物作出用核酸から外来遺伝子を制限酵素で切り出して、再連結したり、上記したトランスジェニック動物作出用核酸の構築方法において、外来遺伝子を含めない方法により容易に構築できる。このような中間体核酸を一旦構築しておけば、この中間体核酸に所望の外来遺伝子を挿入するだけで、所望のトランスジェニック動物作出用核酸を構築することができるので便利である。
【0024】
また、マイクロインジェクション法により注入される核酸断片は、上記のように直鎖状の核酸断片が好ましいが、このような核酸断片を増幅したり、クローニングや精製するために、例えば大腸菌のような宿主細胞中で増殖可能なベクター中に含む組換えベクターを構築すると便利である。本発明は、このような組換えベクターをも提供する。これは、上記した、トランスジェニック動物作出用核酸を構築する方法において、最後にトランスジェニック動物作出用核酸を切り出す前の組換えベクター等であり、上記方法により構築することができる。
【0025】
以下、本発明を実施例に基づきより具体的に説明する。もっとも、本発明は下記実施例に限定されるものではない。
【実施例】
【0026】
1. ベクターの構築
眼の水晶体でマーカーとなるEGFPを発現させるために、図1に示す構造を有するベクター:mαAcrys-globinEGFPを次のようにして構築した。まず、ウサギβ-グロビンのエキソン2〜ポリアデニレーションシグナル部位を含むBamHI-XbaI断片(塩基配列を配列番号6に示す)を調製した。これは、ウサギゲノムDNAを鋳型とし、フォワード側プライマーとしてctgagtgaactgcactgtgac、リバース側プライマーとしてtctagatatgtccttccgagtgagaを用いたPCRにより調製した。なお、リバース側プライマーにはXbaI部位が5'末端に付加してある。PCR ポリメラーゼはPfuTurbo DNA polymerase(商品名、STRATAGENE社)を使用し、「94℃/180秒→(94℃/25秒→72℃/180秒)7サイクル→(94℃/25秒→67℃/180秒)35サイクル→67℃/420秒→4℃/∞」の反応条件で反応を行なった。得られたPCR産物はpCR4Blunt-TOPO(Invitrogen社)にサブクローニングし、その後シークエンスにて配列を確認した。
【0027】
市販のベクターであるpBluescript SK(-)(Stratagene社製)をBamHIとXbaIで処理し、上記で調製したBamHI-XbaI断片を挿入した。そして、ウサギβ-グロビンのエキソン3に存在するEcoRI部位(配列番号6の673nt〜678nt)に、両端にEcoRI認識部位を付加したEGFPのcDNA(733bp)を導入した。なお、両端にEcoRI認識部位を付加したEGFPのcDNAの塩基配列は、配列番号11に示す。このcDNA断片は、次のようにして作製した。EcoRIの認識配列を5'末端に付加したフォワード側プライマーであるEGFP(Eco Kozak-):gaattccgccaccatggtgagcaagとリバース側プライマーであるEGFP(Eco stop-):gaattcttacttgtacagctcgtccを使用し、EGFPのcDNAを含む市販のプラスミドであるpEGFP-N1(クローンテック社)をテンプレートにしてPCRにて調製した。PCRポリメラーゼはPfuTurbo DNA polymerase(商品名、STRATAGENE社)を使用し、「94℃/180秒→(94℃/30秒→50℃/30秒→72℃/60秒)30サイクル→72℃/420秒→4℃/∞」の反応条件で反応を行なった。得られたPCR 産物はpCR4Blunt-TOPO(商品名、Invitrogen社)にサブクローニングし、その後シークエンスにて配列を確認した。最後に、制限酵素EcoRIにより切り出し、EGFPのcDNA(733bp)断片(配列番号11)を作製した。
【0028】
次に、マウスαAクリスタリンのプロモーター(-367〜+51)を含むEcoRV-BamHI断片(塩基配列を配列番号3に示す)を作製した。マウスαAクリスタリンのプロモーターを含有するEcoRV-BamHI断片は、プライマーにそれぞれEcoRVおよびBamHIの認識配列を付加した、フォワード側プライマーmaACrysta-1: gatatcagatctctggaggtttcggag とリバース側プライマーmaACrysta-2: ggatccagggaccgaaggctggcagtgを使用し、マウスゲノムDNAからPCRにて調製した。PCR ポリメラーゼはPfuTurbo DNA polymerase(商品名、STRATAGENE社)を使用し、「94℃/180秒→(94℃/25秒→72℃/180秒)7サイクル→(94℃/25秒→67℃/180秒)35サイクル→67℃/420秒→4℃/∞」の反応条件で反応を行なった。得られたPCR産物はpCR4Blunt-TOPO(商品名、Invitrogen社)にサブクローニングし、その後シークエンスにて配列を確認した。上記のように構築した、pBluescript SK(-)(商品名)を基礎とする組換えベクターをEcoRVとBamHIで消化し、上記EcoRV-BamHI断片を挿入して、図1に示す構造を有する組換えベクターpBSmαAcrys-globinEGFPを得た。
【0029】
2. 供試動物
卵子および精子のドナーとなるB6D2F1マウス、および胚移植レシピエントに用いたICR雌性マウス、偽妊娠誘起交配用精管結紮ICR雄性マウスは全て日本クレア株式会社から購入したものを使用した。これらのマウスは室温22〜24℃、湿度60〜70%、明期と暗期をそれぞれ7:00点灯、19:00消灯となるように設定した飼育環境で飼育した。水、餌はマウスに自由摂取させ、水は水道水を、餌は日本クレア株式会社から購入したCE-2固形飼料(10KGRY放射線滅菌済み)を使用した。なお、購入したマウスは上記の環境に1週間以上馴馳させた後に実験に用いた。B6D2F1マウスは8〜12週齢の個体を用いた。レシピエントに用いたICR雌性マウスは8〜12週齢かつ体重が28〜34gの個体に限定した。
【0030】
3. 前核期卵の採卵
8〜12週令のB6D2F1雌性マウスに7.5IUの妊馬血清性性腺刺激ホルモン(Pregnant Mare's Serum Gonadotropin:PMSG(帝国臓器製薬株式会社))を腹腔内に注射し、47〜48時間後に8.25IUのヒト絨毛性性腺刺激ホルモン(Human Chorionic Gonadotropin:hCG(デンカ製薬株式会社))を同じく腹腔内に注射し、過排卵を誘起した。その後交配用B6D2F1雄性マウスのケージで一晩交配させ、翌朝膣栓(プラグ)が確認できた雌性マウスを前核期卵のドナーマウスとした。hCG注射後20〜21時間でドナーマウスを頚椎脱臼により安楽死させた後、卵管を摘出し、実体顕微鏡下で卵管膨大部の切り裂くことにより卵丘細胞卵子複合体を採取した。卵丘細胞卵子複合体は、ヒアルロニダーゼ(SIGMA CHEMICAL CO.) (CZB培地に25倍濃度で溶解した状態で−20℃保存)処理(750〜1500unit/ml)によって卵丘細胞を取り除いた後、雌雄両前核の形成が確認され、かつ形態学的に正常な卵子のみを選別し、実験の供試卵とした。なお、CZB培地の組成は次の通りである。NaCl 81.62mM、KCl 4.83mM、MgSO4・7H2O 1.18mM、KH2PO4 1.18mM、EDTA・2Na 0.11mM、Na-Lactate 19.76mM、D-Glucose 5.55mM、CaCl2・2H2O 1.7mM、NaHCO3 25.12mM、ピルビン酸ナトリウム 0.27mM、L-グルタミン 1.00mM、BSA 5.00mg/ml、ペニシリン G 75mg/L, ストレプトマイシン 50mg/L
【0031】
4. トランスジェニック動物作出用核酸の調製
上記1で構築した、図1に示す構造を有する組換えベクターpBSmαAcrys-globinEGFPをKpnIとNotIで消化して2つに切断し、消化物をアガロースゲル電気泳動にかけて2つの断片を分離した。遺伝子導入に用いるKpnI(653)からNotI(2717)の断片(図1参照)をアガロースゲルから切り出し、GENECLEAN(商品名)にて精製して、トランスジェニック動物作出用核酸を得た。これを、pH7.5に調整したTE bufferで10ng/μl濃度に希釈して、後述する前核へのマイクロインジェクションに用いた。
【0032】
5. 前核注入法
前核注入法については非特許文献1の方法を参考にした。前核注入法によって染色体上に組み込まれた外来DNAは、1〜数100コピーの長さで1〜複数の遺伝子座のランダムな位置に整列した状態で組み込まれるという報告がなされている。(非特許文献2) そのため注入に用いるプラスミドDNAは環状のままではなく、制限酵素を用いて直鎖状にしたものを用いたほうがその導入効率は向上し、現在でも制限酵素によるプラスミドDNAの切断処理は広く支持されている。前核注入はM2培地 (SIGMA社製、組成:CaCl2・2H2O 0.251g/L、MgSO4 0.165g/L、KCl 0.356g/L、KH2PO4 0.162g/L、NaCl 5.532g/L、NaHCO3 0.35g/L、アルブミン(Bovine FractionV) 4.0g/L、D-グルコース 1.0g/L、HEPES 5.43g/L、フェノールレッドNa塩 0.01g/L、ピルビン酸ナトリウム 0.036g/L、乳酸ナトリウム 4.35g/Lにペニシリン G 60mg/L、ストレプトマイシン 50mg/Lを添加)を用いて行った。前核注入法は、より具体的には次のようにして行なった。
【0033】
ホールディングピペットはガラスキャピラリー(CLARK ELECTROMEDICAL INSTRUMENT GC100-15)をアルコールランプで熱して引き、外径が約100μmの位置をルビーカッターにより切断した。さらに、ガラスキャピラリーをマイクロフォージ(NARISHIGE CO.JAPAN MF-79)のフィラメント上のガラス玉に垂直になるようにセットし、フィラメントを熱して内径が約10μmとなるようにガラスキャピラリー断面を丸めた。作製したホールディングピペットは乾熱滅菌し、使用時まで滅菌保存した。また、インジェクションピペットは乾熱滅菌しておいたガラスキャピラリー(CLARK ELECTROMEDICAL INSTRUMENT GC100TF-15)をキャピラリープラー(SUTTER INSTRUMENT Co. P-97/IVF)を用いて作製した。
【0034】
倒立顕微鏡ステージ上のホットプレートを37.5℃に設定し、ミネラルオイル中に5μlのM2培地ドロップが6〜8個になるように作製したマニピュレーション用ディッシュをステージに乗せた。ホールディングピペットをマイクロインジェクター(NARISHIGE CO.JAPAN IM9B)に、DNA溶液を充填したインジェクションピペットをインジェクター(NARISHIGE CO.JAPAN IM−200)にセットし、両ピペットの先端がM2培地ドロップ内に位置するようにセットした。本発明では、DNA溶液としてKpnI/NotI処理により直鎖状にしたmαAcrys-globinEGFPをpH7.5に調整したTE bufferで10 ng/μl濃度に希釈し、インジェクションピペットに充填した。
【0035】
マニピュレーション用ディッシュ中のM2培地ドロップ内へ20〜30個の受精卵を入れ、インジェクションピペットを受精卵の雄性前核に刺し、上記4で調製した、mαAcrys-globinEGFP由来のトランスジェニック動物作出用核酸溶液を約2pl注入した。M2培地ドロップ内の受精卵へ注入を終えたらCZB培地に受精卵を移し、20〜30分間インキュベータ内で培養して生存判定後、生存卵のみを桑実期胚まで培養した。培養は37.5℃、5%CO2 in air、湿度100%の条件に設定された炭酸ガス培養装置内で行なった。
【0036】
6. レシピエントマウスへの胚移植
桑実期胚まで発生した胚を胚移植に用いた。レシピエントマウスには8〜12週令のICR雌性を用いた。陰部の観察によって発情前期であると確認できたICR雌性マウスを、精管結紮処置を施したICR雄性マウスのケージで一晩交配させ、翌朝プラグが確認できた偽妊娠マウスをレシピエントとして用いた。プラグが確認できた時点を交配0.5日後(0.5 days post coitus :0.5 d.p.c.)とし、桑実期胚は3 d.p.c.の偽妊娠ICR雌性マウス子宮へ移植した。その後19〜20d.p.c.でレシピエントマウスを剖検し、胎仔を回収した。
【0037】
7. トランスジェニックマウスの判定
19〜20d.p.c.で回収した胎仔を解剖して眼を取り出し、実体蛍光顕微鏡にて視軸方向に沿って角膜より水晶体を観察した。観察した全32匹の胎仔のうち、7匹の胎仔の水晶体が蛍光を発していた。また、他の組織についても同様に観察したところ、蛍光を発している組織は観察されなかった。これらのマウスよりゲノムDNAを分離し、PCRにてマーカー遺伝子の確認を行なったところ、EGFP発現ベクター:mαAcrys-globinEGFPが導入されたトランスジェニックマウスと蛍光を発光していたマウスが一致した。
【0038】
8. 組織特異的発現の確認
トランスジェニックマウスと確認されたマウスについて、眼、内臓塊おけるEGFPの発現をRT-PCRで確認した。これは具体的に次のようにして行なった。仮親マウスから摘出したマウス胎児から眼球を摘出して眼組織とし、胸腔及び腹腔内に存在する臓器をまとめて内臓塊として処理した。それぞれの組織にISOGEN(商品名、ニッポンジーン社製)を加えてホモジナイズし、室温にて5分放置後、クロロホルムを加えて12000G、4℃、15分間遠心分離した。二層に分かれた水層(上層)を別のチューブに移し、イソプロピルアルコールを加えてイソプロピルアルコール沈殿を行い、沈殿としてtotal RNAを回収した。Total RNAはDEPC処理水に溶解し、100ng/μlの濃度に調製した。PCRチューブに総RNA 100ng、oligo(dT12-18) 0.5μg、10mM dNTP mix 1μlを加え、DEPC処理水により全量を12μlとした。70℃にて10分間インキュベーションし、すばやく氷上に移した。さらに5X first strand buffer 4μl(Invitrogen社)、0.1M DTT 2μl、RNaseOUT 1μl(商品名、Invitrogen社)を加え、全量を19μlとした。42℃、1分間のプレインキュベーション後、スーパースクリプトIIRNaseH-逆転写酵素(Invitrogen社)を1μl加え、42℃ 50分間インキュベーションした。最後に70℃ 15分間インキュベーションして反応を止め、Ribonuclease H(タカラバイオ社)を加えて第1鎖cDNAを完成させた。
【0039】
得られた第1鎖cDNAをテンプレートとし、フォワード側プライマーにRTRab-b-GB(Ex.2):ggatcctgagaacttcagg、リバース側プライマーにRTEGFP:agtcgtgctgcttcatgtgを使用してPCRを行なった。PCR ポリメラーゼはTaKaRa Ex Taq(タカラバイオ社)を使用し、「94℃/180秒→(94℃/25秒→72℃/180秒)7サイクル→(94℃/25秒→67℃/180秒)32サイクル→72℃/420秒→4℃/∞」の反応条件で反応を行なった。ゲノムに取り込まれたmαAcrys-globinEGFPがテンプレートになった場合は903bpのPCRプロダクトが得られる。また、mαAcrys-globinEGFPから転写されたmRNAはスプライシングを受けるので、mRNA由来のcDNAがテンプレートになった場合は330bpのPCRプロダクトが確認される。今回のRT-PCRの結果、トランスジェニックマウスの眼球由来mRNAから330bpのPCRプロダクトが得られたので、眼球特異的にEGFP mRNAの転写が行なわれていることが確認された。この結果、EGFP遺伝子は眼特異的に働いていることが確認され、それ以外の組織における発現は確認されなかった。
【図面の簡単な説明】
【0040】
【図1】実施例において作製した、マウスαAプロモーターと、EGFP遺伝子を含む組換えベクターの遺伝子地図である。
【図2】外来遺伝子としてアジポネクチン遺伝子を導入するために用いられる組換えベクターの遺伝子地図である。
【特許請求の範囲】
【請求項1】
所望の外来遺伝子と、宿主細胞内で該外来遺伝子の発現を制御するプロモーターを含む核酸を受精卵若しくはクローン卵又は胚に導入し、該受精卵若しくはクローン卵又は胚から個体を発生させるトランスジェニック動物の作出方法において、水晶体において特異的に発現する遺伝子のプロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される、可視的に発現の有無を判定できるマーカータンパク質をコードするマーカー遺伝子を、前記核酸に連結して導入することを特徴とする、トランスジェニック動物の作出方法。
【請求項2】
前記水晶体において特異的に発現する遺伝子のプロモーターが、αAクリスタリンのプロモーターである請求項1記載の方法。
【請求項3】
前記核酸を、受精卵の前核にマイクロインジェクションすることにより前記外来遺伝子を導入する請求項3記載の方法。
【請求項4】
前記マーカー遺伝子が、蛍光タンパク質をコードする遺伝子である請求項1ないし4のいずれか1項に記載の方法。
【請求項5】
前記蛍光タンパク質が、GFP、EGFP、EBFP、ECFP、EYFP、AmCyan1、ZsGreen1、ZaYellow1、DsRed2、AsRed2及びHcRed1から成る群より選ばれる少なくとも1種である請求項5記載の方法。
【請求項6】
前記マーカー遺伝子の下流及び前記外来遺伝子の下流にそれぞれターミネーターを含む核酸を導入する請求項1ないし5のいずれか1項に記載の方法。
【請求項7】
請求項1ないし6のいずれか1項に記載の方法により作出され、水晶体内において前記マーカー遺伝子を特異的に発現しているトランスジェニック動物又は水晶体内において前記マーカー遺伝子を特異的に発現するその子孫。
【請求項8】
水晶体において特異的に発現する遺伝子のプロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される、可視的に発現の有無を判定できるタンパク質をコードするマーカー遺伝子と、所望の外来遺伝子と、該外来遺伝子の上流に位置し、該外来遺伝子の発現を制御するプロモーターとを含む、トランスジェニック動物作出用核酸。
【請求項9】
水晶体において特異的に発現する遺伝子のプロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される、可視的に発現の有無を判定できるタンパク質をコードするマーカー遺伝子と、所望の外来遺伝子を挿入するための制限酵素部位と、該制限酵素部位の上流に位置し、該制限酵素部位に挿入される外来遺伝子の発現を制御するプロモーターとを含む、請求項8記載の核酸を構築するための核酸。
【請求項10】
前記水晶体において特異的に発現する遺伝子のプロモーターがαAクリスタリンのプロモーターである請求項8又は9記載の核酸。
【請求項11】
前記マーカー遺伝子が、蛍光タンパク質をコードする遺伝子である請求項8ないし9のいずれか1項に記載の核酸。
【請求項12】
前記蛍光タンパク質が、GFP、EGFP、EBFP、ECFP、EYFP、AmCyan1、ZsGreen1、ZaYellow1、DsRed2、AsRed2及びHcRed1から成る群より選ばれる少なくとも1種である請求項11記載の核酸。
【請求項13】
前記マーカー遺伝子の下流及び前記外来遺伝子の下流にそれぞれターミネーターを含む請求項8ないし12のいずれか1項に記載の核酸。
【請求項14】
請求項8ないし13のいずれか1項に記載の核酸と、宿主細胞中での複製を可能にする複製開始点を少なくとも含む組み換えベクター。
【請求項1】
所望の外来遺伝子と、宿主細胞内で該外来遺伝子の発現を制御するプロモーターを含む核酸を受精卵若しくはクローン卵又は胚に導入し、該受精卵若しくはクローン卵又は胚から個体を発生させるトランスジェニック動物の作出方法において、水晶体において特異的に発現する遺伝子のプロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される、可視的に発現の有無を判定できるマーカータンパク質をコードするマーカー遺伝子を、前記核酸に連結して導入することを特徴とする、トランスジェニック動物の作出方法。
【請求項2】
前記水晶体において特異的に発現する遺伝子のプロモーターが、αAクリスタリンのプロモーターである請求項1記載の方法。
【請求項3】
前記核酸を、受精卵の前核にマイクロインジェクションすることにより前記外来遺伝子を導入する請求項3記載の方法。
【請求項4】
前記マーカー遺伝子が、蛍光タンパク質をコードする遺伝子である請求項1ないし4のいずれか1項に記載の方法。
【請求項5】
前記蛍光タンパク質が、GFP、EGFP、EBFP、ECFP、EYFP、AmCyan1、ZsGreen1、ZaYellow1、DsRed2、AsRed2及びHcRed1から成る群より選ばれる少なくとも1種である請求項5記載の方法。
【請求項6】
前記マーカー遺伝子の下流及び前記外来遺伝子の下流にそれぞれターミネーターを含む核酸を導入する請求項1ないし5のいずれか1項に記載の方法。
【請求項7】
請求項1ないし6のいずれか1項に記載の方法により作出され、水晶体内において前記マーカー遺伝子を特異的に発現しているトランスジェニック動物又は水晶体内において前記マーカー遺伝子を特異的に発現するその子孫。
【請求項8】
水晶体において特異的に発現する遺伝子のプロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される、可視的に発現の有無を判定できるタンパク質をコードするマーカー遺伝子と、所望の外来遺伝子と、該外来遺伝子の上流に位置し、該外来遺伝子の発現を制御するプロモーターとを含む、トランスジェニック動物作出用核酸。
【請求項9】
水晶体において特異的に発現する遺伝子のプロモーターと、該プロモーターの下流に位置し、該プロモーターにより制御される、可視的に発現の有無を判定できるタンパク質をコードするマーカー遺伝子と、所望の外来遺伝子を挿入するための制限酵素部位と、該制限酵素部位の上流に位置し、該制限酵素部位に挿入される外来遺伝子の発現を制御するプロモーターとを含む、請求項8記載の核酸を構築するための核酸。
【請求項10】
前記水晶体において特異的に発現する遺伝子のプロモーターがαAクリスタリンのプロモーターである請求項8又は9記載の核酸。
【請求項11】
前記マーカー遺伝子が、蛍光タンパク質をコードする遺伝子である請求項8ないし9のいずれか1項に記載の核酸。
【請求項12】
前記蛍光タンパク質が、GFP、EGFP、EBFP、ECFP、EYFP、AmCyan1、ZsGreen1、ZaYellow1、DsRed2、AsRed2及びHcRed1から成る群より選ばれる少なくとも1種である請求項11記載の核酸。
【請求項13】
前記マーカー遺伝子の下流及び前記外来遺伝子の下流にそれぞれターミネーターを含む請求項8ないし12のいずれか1項に記載の核酸。
【請求項14】
請求項8ないし13のいずれか1項に記載の核酸と、宿主細胞中での複製を可能にする複製開始点を少なくとも含む組み換えベクター。
【図1】


【図2】
【図2】
【公開番号】特開2007−82446(P2007−82446A)
【公開日】平成19年4月5日(2007.4.5)
【国際特許分類】
【出願番号】特願2005−273724(P2005−273724)
【出願日】平成17年9月21日(2005.9.21)
【国等の委託研究の成果に係る記載事項】(出願人による申告)国等の委託研究の成果に係る特許出願(平成17年度生物系特定産業技術研究支援センター「新技術・新分野創出のための基礎研究推進事業」、産業活力再生特別措置法第30条の適用を受けるもの)
【出願人】(301000505)株式会社バイオス医科学研究所 (10)
【出願人】(801000027)学校法人明治大学 (161)
【Fターム(参考)】
【公開日】平成19年4月5日(2007.4.5)
【国際特許分類】
【出願日】平成17年9月21日(2005.9.21)
【国等の委託研究の成果に係る記載事項】(出願人による申告)国等の委託研究の成果に係る特許出願(平成17年度生物系特定産業技術研究支援センター「新技術・新分野創出のための基礎研究推進事業」、産業活力再生特別措置法第30条の適用を受けるもの)
【出願人】(301000505)株式会社バイオス医科学研究所 (10)
【出願人】(801000027)学校法人明治大学 (161)
【Fターム(参考)】
[ Back to top ]