
ヒト型糖鎖をもつ糖タンパク質の生産方法
【課題】ヒト型の糖鎖をもつ有用糖タンパク質を提供すること。
【解決手段】ヒト型糖鎖をもつ糖タンパク質の生産方法であって、植物細胞に、グリコシルトランスフェラーゼの遺伝子および異種糖タンパク質の遺伝子を導入することによって、形質転換植物細胞を得る工程、および得られた形質転換植物細胞を培養する工程を包含する。上記グリコシルトランスフェラーゼは、非還元末端アセチルグルコサミン残基へのガラクトース残基の転移反応を行い得る。上記ヒト型糖鎖をもつ糖タンパク質は、コア糖鎖および外部糖鎖を含み、このコア糖鎖は複数のマンノースおよびアセチルグルコサミンを含み、そしてこの外部糖鎖は、非還元末端ガラクトースをもつ末端糖鎖部分を含み、直鎖状構造または分岐状構造をもつ。上記ヒト型糖鎖をもつ糖タンパク質は、フコースまたはキシロースを含まないため、ヒトに対する抗原性を示さない。
【解決手段】ヒト型糖鎖をもつ糖タンパク質の生産方法であって、植物細胞に、グリコシルトランスフェラーゼの遺伝子および異種糖タンパク質の遺伝子を導入することによって、形質転換植物細胞を得る工程、および得られた形質転換植物細胞を培養する工程を包含する。上記グリコシルトランスフェラーゼは、非還元末端アセチルグルコサミン残基へのガラクトース残基の転移反応を行い得る。上記ヒト型糖鎖をもつ糖タンパク質は、コア糖鎖および外部糖鎖を含み、このコア糖鎖は複数のマンノースおよびアセチルグルコサミンを含み、そしてこの外部糖鎖は、非還元末端ガラクトースをもつ末端糖鎖部分を含み、直鎖状構造または分岐状構造をもつ。上記ヒト型糖鎖をもつ糖タンパク質は、フコースまたはキシロースを含まないため、ヒトに対する抗原性を示さない。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、植物による異種糖タンパク質の発現に関する。
【背景技術】
【0002】
生体内の機能的タンパク質の多くは糖鎖を持つ糖タンパク質である。糖タンパク質内の糖鎖の多様性は、いくつかの重要な役割を生理学的に担っていることが明らかにされている(Lain,R.A.,Glycobiology,4,759−767,1994)。
【0003】
最近、糖鎖の作用が2つのカテゴリーに分けられ得ることが明確になった。1つは、糖鎖が、血液からの糖タンパク質のクリアランス、リソソーム酵素のリソソームへのターゲッティング、糖タンパク質による特定の組織および器官へのターゲッティングにおいて、細胞間接着のリガンドとして、および細菌およびウイルスの受容体として直接的な機能を有する場合である。例えば、エイズウイルス(HIV)が標的細胞に感染する過程で、糖タンパク質の糖鎖が関与していることが示されている(Rabehi,L.ら、Glycoconj.J.,12,7−16,1995)。HIV表面は、エンベロープタンパク質gp120で覆われ、gp120の糖鎖が標的細胞のCD4に結合することでHIVウイルスの感染が始まる。糖鎖のもう1つの働きは、糖鎖自体は機能的分子ではないが、タンパク質の高次構造の形成、タンパク質の溶解性、タンパク質のプロテアーゼ抵抗性、抗原性の抑制、タンパク質機能の修飾、タンパク質代謝速度の調節および細胞膜における発現量の調節などに間接的に関与する場合である。例えば、神経系に広く分布する神経細胞接着分子の接着性は糖鎖により調節されている(Edelman,G.M.,Ann.Rev.Biochem.,54,135−169,1985)。
【0004】
真核生物では、糖タンパク質の糖鎖は、小胞体の脂質上で前駆体型糖鎖として合成される。糖鎖部分はタンパク質に転移され、次いでこのタンパク質上のいくつかの糖残基が除去され、ゴルジ体に輸送される。ゴルジ体において、過剰の糖残基が除去された後、さらなる糖残基(例えばマンノース)が付加され、そして糖鎖が伸長される(Narimatsu,H.,Microbiol.Immunol.,38,489−504,1994)。
【0005】
より詳細に述べれば、例えば、ドリコールアンカー(dolichol anchors)上のGlc3Man9GlcNAc2は、小胞体(ER)膜中のタンパク質に転移される(Moremen K.W.,Trimble、R.B.およびHerscovics A.,Glycobiology 1994 4月;4(2):113−25、アスパラギン連結オリゴサッカライドプロセッシング経路のグリコシダーゼ;およびSturm,A.1995 植物タンパク質のN−グリコシル化、New Comprehensive Biochemistry.Glycoproteins,第29a巻、Montreuil、J.,Schachter、H.およびVliegenthart、J.F.G.(編).Elsevier Science Publishers B.V.,The Netherland、521−541頁)。小胞体グリコシダーゼIおよびIIは、3つのグルコース単位を除去する(Sturm、A.1995、前述;およびKaushal G.P.およびElbein A.D.,1989、植物における糖タンパク質プロセッシング酵素。Methods Enzymology 179、Complex Carbohydrates Part F.Ginsburg V.(編)、Academic Press,Inc.NY、452−475頁)。得られる高マンノース構造(Man9GlcNAc2)は、小胞体マンノシダーゼによりトリミングされる(Moremen K.W.ら、前述;およびKornfeld,R.およびKornfeld,S.,Annu.Rev.Biochem.54、631−664、1985;アスパラギン連結オリゴサッカライドのアセンブリ)。除去されるマンノース残基の数は、プロセッシング酵素への接近可能性における差異に従って変動する。アイソマー類であるMan8−、Man7−、Man6−およびMan5GlcNAc2が、小胞体マンノシダーゼおよびマンノシダーゼIによるプロセッングの間に生産される(Kornfeld,R.およびKornfeld,S.,前述)。4つのマンノース残基がマンノシダーゼI(Man I)によって完全に除去されるとき、産物は、Man5GlcNAc2である。N−アセチルグルコサミニルトランスフェラーゼI(GlcNAcI)は、N−アセチルグルコサミン(GlcNAc)を、UDP−GlcNAcからMan5GlcNAc2へ転移し、GlcNAcMan5GlcNAc2を生じる(Schachter,H.,Narasimhan,S.,Gleeson,P.,およびVella,G.,グリコシルトランスフェラーゼは、複合型またはN−アセチルガラクトサミン型のN−グリコシル的に連結されたオリゴサッカライドの伸長に関与する。Methods Enzymol 98:Biomembranes Part L.Fleischer,S.,およびFleischer,B.(編)、Academic Press,Inc.NY.98−134頁、1983)。マンノシダーゼII(Man II)は、GlcNAcMan5GlcNAc2から2つのマンノース残基を除去し、GlcNAcMan3GlcNAc2を生じる(Kaushal,G.P.およびElbein,A.D.,前述;およびKornfeld,R.およびKornfeld,S.,前述)。オリゴサッカライドGlcNAcMan4GlcNAc2は、N−アセチルグルコサミニルトランスフェラーゼII(GlcNAcII)の基質として用いられる(Moremen,K.W.ら、前述;Kaushal,G.P.およびElbein,A.D.,前述;およびKornfeld,R.およびKornfeld,S.,前述)。図19は、N−結合グリカンの上記の構造、ならびに小胞体およびゴルジ体中の糖鎖修飾経路に関与する酵素を要約する。図19において、白抜きの菱形はグルコースを、白抜きの四角はGlcNAcを、白抜きの丸はマンノースを、黒丸はガラクトースを、そして黒四角はシアル酸をそれぞれ示す。
【0006】
このゴルジ体における糖付加は末端部糖鎖合成と呼ばれ、このプロセスは、生物種により大きく異なる。糖鎖生合成系は、真核生物の種によって異なり、その結果糖鎖構造は種特異的であり、そして糖を付加する糖転移酵素およびゴルジ体の進化を反映している(成松久、細胞工学、15,802−810,1996)。
【0007】
アスパラギン結合型(N−結合型)糖鎖に注目すると、動物には、高マンノース型糖鎖、複合型糖鎖およびそのハイブリッド型が存在する。図1にそれらの構造を示す。一方、植物の複合型糖鎖には、動物の糖鎖にはないα1−3フコースとβ1−2キシロースが存在する(Johnson,K.D.およびChrispeels,M.J.,Plant Physiol,84,1301−1308,1897、Kimura,Y.ら,Biosci.Biotech.Biochem.,56,215−222,1992)。また、N結合型糖鎖としては植物糖鎖には動物糖鎖に見出されるシアル酸が存在せず、動物糖鎖に一般に存在するガラクトースも、シカモア細胞のラッカーゼの糖鎖に見出されているが(Takahashi,N.およびHotta,T.ら,Biochemistry,25,388−395,1986)、その例は少なく、またその結合型もβ1−3結合である(FEBS Lett 1997 9月 29,415(2)、186−191、植物細胞懸濁培養(Vaccinium mytillus L.)からの分泌ペルオキシダーゼ中のヒトLewis(a)炭水化物モチーフの同定、Melo NS、Nimtz M、Contradt HS、Fevereiro PS、Costa J;Plant J.1997 12月.12(6)1411−1417、Lewis aエピトープを保持するN−グリカンは、植物細胞の表面で発現される。Fitchette−Laine AC、Gomord V、Cabanes M、Michalski JC、Saint Macary M、Foucher B、Cavelier B、Hawes C、Lerouge P、Faye L)。この結合は、動物で見出される結合とは異なる。
【0008】
一般に、ヒトエリスロポエチン(EPO)などヒト由来の有用な糖タンパク質は、ヒトに近い糖鎖構造を有する糖タンパク質を生産する動物細胞宿主で生産されている。しかし、動物細胞で生産されたEPOは、本来の糖鎖構造とは異なる糖鎖構造を有し、生体内における活性が低下した(Takeuchi,M.ら,Proc.Natl.Acad.Sci.USA,86,7819−7822,1989)。ホルモン、インターフェロンなどのヒト由来の他の有用タンパク質の糖鎖構造が解析され、EPOと同じグルコシル化制限をともなって工業的に生産されている。
【0009】
近年、外来遺伝子を植物に導入する方法として、アグロバクテリウム法(Weising,K.ら,Annu.Rev.Genet.,22,421,1988)、エレクトロポレーション法(Toriyama,K.ら,Bio/Technology,6,1072,1988)、金粒子法(Gasser,C.G.およびFraley,R.T.、Science,244,1293,1989)などが確立され、アルブミン(Sijmons,P.C.ら,Bio/Technology,8,217,1990)、エンケファリン(Vandekerckhove,J.ら,Bio/Technology,7,929,1989)、およびモノクローナル抗体(Benvenulo,E.ら,Plant Mol.Biol.,17,865,1991およびHiatt,A.ら,Nature,342,76,1989)、が植物で生産され、そしてB型肝炎ウイルス主要表面抗原(HBsAg)(Mason,H.S.ら,Proc.Natl.Acad.Sci.USA.,89,11745,1992)、および分泌型IgA(Hiatt,A.およびMa,J.S.K.,FEBS Lett.,307,71,1992)を植物細胞中で生産し得ることも記載されている。しかし、ヒト由来の糖タンパク質を植物で発現する場合、植物は、ヒトと異なる糖鎖付加機構を有するため、生産される糖タンパク質の糖鎖は、ヒトで生産される糖鎖とは異なる構造を有し、本来の生理活性を示さず、また抗原となり得ることが指摘されている(Wilson I.B.H.ら,Glycobiol.,vol.8,No.7,pp651−661,1998)。
【発明の概要】
【課題を解決するための手段】
【0010】
本発明の目的は、哺乳動物、例えば、ヒト型の糖鎖をもつ植物が生産する組換え糖タンパク質を提供することにより、上記先行技術に関連する課題を解決することである。
【0011】
本発明は、ヒト型糖鎖をもつ糖タンパク質の生産方法に関する。この方法は、植物細胞に、非還元末端アセチルグルコサミン残基へのガラクトース残基の転移反応を行い得る酵素の遺伝子および異種糖タンパク質の遺伝子を導入して形質転換細胞を得る工程、および得られた形質転換植物細胞を培養する工程を包含する。
【0012】
本発明において、上記ヒト型糖鎖をもつ糖タンパク質は、コア糖鎖および外部糖鎖を含み、上記コア糖鎖は、複数のマンノースおよびアセチルグルコサミンから本質的になり、上記外部糖鎖は、非還元末端ガラクトースを含む末端糖鎖部分を含み得る。
【0013】
本発明において、上記外部糖鎖は、直鎖状構造または分岐状構造を有し得る。
【0014】
本発明において、上記分岐糖鎖部分は、モノ、バイ、トリ、またはテトラ構造であり得る。
【0015】
本発明において、上記糖タンパク質は、フコースおよびキシロースを含まなくともよい。
【0016】
本発明はまた、1つの局面で、非還元末端アセチルグルコサミン残基へのガラクトース残基の転移反応を行い得る糖鎖付加機構を備えた植物細胞に関し、この糖鎖付加機構は、コア糖鎖および外部糖鎖を含む糖鎖であって、このコア糖鎖は、複数のマンノースおよびアセチルグルコサミンから本質的になり、この外部糖鎖は非還元末端ガラクトースを含む末端糖鎖部分を含む糖鎖を付加する。
【0017】
本発明はまた、1つの局面で上記の方法によって得られたヒト型糖鎖をもつ糖タンパク質に関する。
【図面の簡単な説明】
【0018】
【図1】代表的なN結合型糖鎖の構造を示す模式図である。
【図2】hGTのクローニングの方法の模式図である。
【図3】hGT発現用ベクターpGAhGTの構築方法の模式図である。
【図4】形質転換体タバコ培養細胞のゲノムのサザン解析を示す写真である。図4(A)は、ゲノムDNA(40μg)をEcoRIおよびHindIIIで消化した後の電気泳動を示す。左側の数字はDNA分子量マーカーの位置を表す。図4(B)は、各形質転換細胞に組み込まれた、プロモーター、hGT、ターミネーターを含む2.2kbの断片の模式図を示す。
【図5】図5は、形質転換タバコBY2細胞(WT)および野生型タバコBY2細胞(WT)からの免疫反応性タンパク質のウェスタンブロッティングの写真である。タンパク質を変性させ、10% SDS−PAGEで電気泳動し、そしてニトロセルロース膜に電気的に転写した。レーンの数字は、サンプルがそれぞれ以下のものに由来したことを示す:1=GT1の細胞抽出物;2=GT6の細胞抽出物;3=GT8の細胞抽出物;4=GT9の細胞抽出物;5=野生型の細胞抽出物;6=GT1のミクロソーム画分;7=GT6のミクロソーム画分;8=GT8のミクロソーム画分;9=GT9のミクロソーム画分;および10=野生型のミクロソーム画分。
【図6】Ricinus communis(RCA120)アフィニティークロマトグラフィーを用いたガラクトシル化糖タンパク質の検出を示す電気泳動写真である。電気泳動ゲルは、銀染色により可視化した。レーン1および2は野生型BY2細胞からのタンパク質を示し、そしてレーン3および4は、形質転換GT6細胞からのタンパク質を示す。分子量はKDaの単位である。
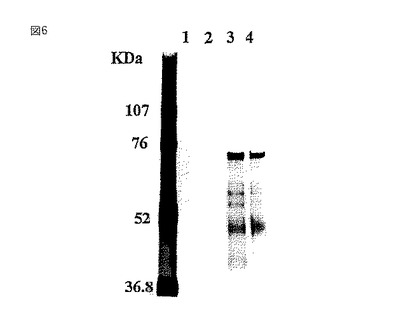
【図7】Ricinus communis(RCA120)アフィニティークロマトグラフィーを用いたガラクトシル化糖タンパク質の検出を示すウェスタンブロッティングの写真である。電気泳動したゲルをニトロセルロース膜上へブロットした後、このブロットをレクチン(RCA120)染色して可視化した。レーン1および2は野生型BY2細胞からのタンパク質を示し、そしてレーン3および4は、形質転換GT6細胞からのタンパク質を示す。分子量はKDaの単位である。
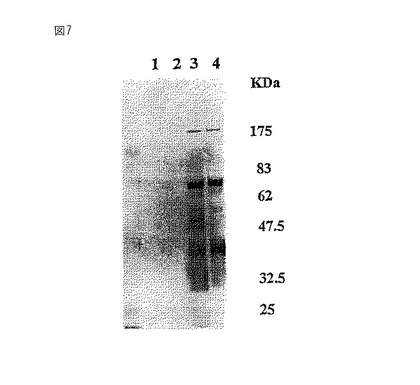
【図8】Ricinus communis(RCA120)アフィニティークロマトグラフィーを用いたガラクトシル化糖タンパク質を、植物の複合型グリカンに存在するキシロースに特異的な抗血清でプローブしたブロッティングの写真である。レーン1および2は、BY2およびGT6からの総タンパク質抽出物をそれぞれ示し、レーン3は、RCA120アフィニティークロマトグラフィー後のGT6からの糖タンパク質を示す。分子量はKDaの単位である。
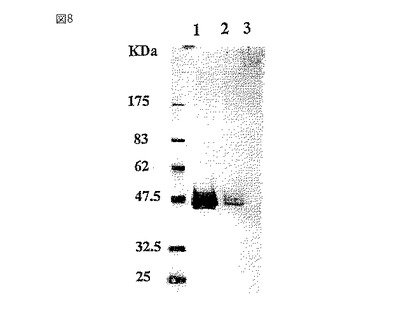
【図9】カナマイシン耐性遺伝子およびハイグロマイシン耐性遺伝子を有するバイナリーベクターであり、HRP cDNAを有するプラスミドpBIHm−HRPの模式図である。
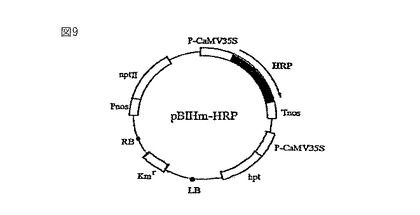
【図10】トランスジェニック懸濁培養細胞におけるHRPの産生を示す等電点電気泳動およびウエスタンブロッティングの写真である。図10(A)は等電点電気泳動の結果を示し、そして図10(B)はウエスタンブロッティングの結果を示す。略語は以下の通りである:WT=野生型;BY2−HRP 1、5、および7=HRP遺伝子で形質転換したBY2細胞のクローン番号;ならびにGT6−HRP 4、5、および8=HRP遺伝子で形質転換したGT6細胞のクローン番号。
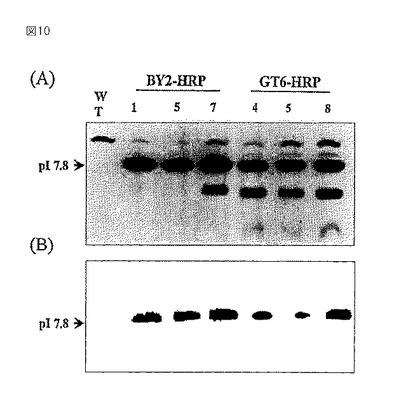
【図11】0.02%TFA中でアセトニトリルの濃度を0〜15%の直線状勾配で60分間かけて増加させ、1.2ml/分の流速で溶出したPA糖鎖の逆相HPLCパターンを示すグラフである。I〜XIは、サイズ分画HPLCにおいて溶出および精製した個々の画分を示す。励起波長および放出波長は、それぞれ、310nmおよび380nmであった。
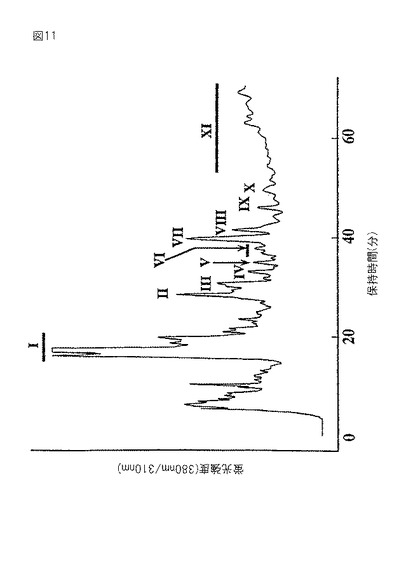
【図12】図11におけるPA鎖のサイズ分画HPLCパターンを示すグラフである。PA糖鎖を、水−アセトニトリル混合物中で水の濃度を30〜50%に40分間かけて増加させ、0.8ml/分の流速で溶出させた。励起波長および放出波長は、それぞれ、310nmおよび380nmであった。
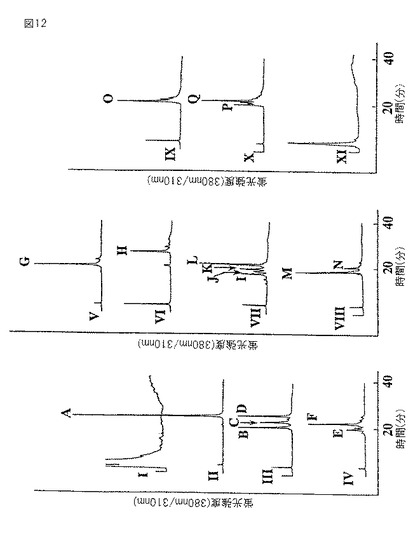
【図13】2つの標準品糖鎖AおよびBと比較した、逆相HPLCにおけるピーク−K2の溶出位置を示すグラフである。溶出条件は、図11と同様に0.02%TFA中でアセトニトリルの濃度を0〜15%の直線状勾配で60分間かけて増加させ、1.2ml/分の流速で溶出であった。
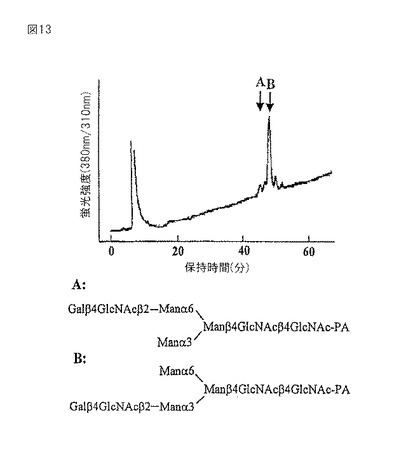
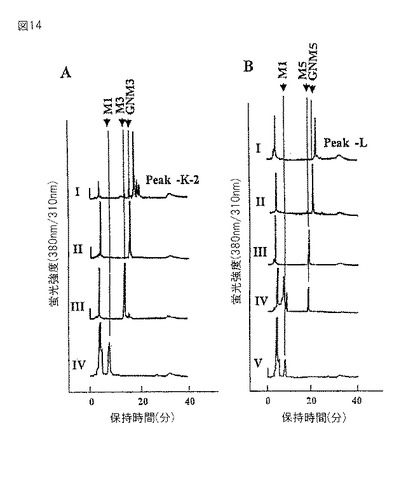
【図14】エクソグリコシダーゼ消化後に得られたガラクトシル化PA糖鎖のSF−HPLCプロフィールを示すグラフである。PA糖鎖を、水−アセトニトリル混合物中の水含量を30〜50%で25分間かけて増加させ、0.8ml/分の流速で溶出させた。(A)PA−糖鎖K−2;I.用いたガラクトシル化PA糖鎖の溶出位置;II.Iのβ−ガラクトシダーゼ消化物;III.IIのN−アセチル−β−D−グルコサミニダーゼ消化物;IV.IIIのタチナタマメ α−マンノシダーゼ消化物。(B)PAー糖鎖L;I.用いたガラクトシル化PA糖鎖の溶出位置;II.Iのβ−ガラクトシダーゼ消化物;III.IIのN−アセチル−β−D−グルコサミニダーゼ消化物;IV.IIIのα1−2マンノシダーゼ消化物;V.IIIのタチナタマメ α−マンノシダーゼ消化物。
【図15】形質転換細胞から得られたN結合型グリカンの推定構造を示す図である。括弧内の数字は、モル比を示す。
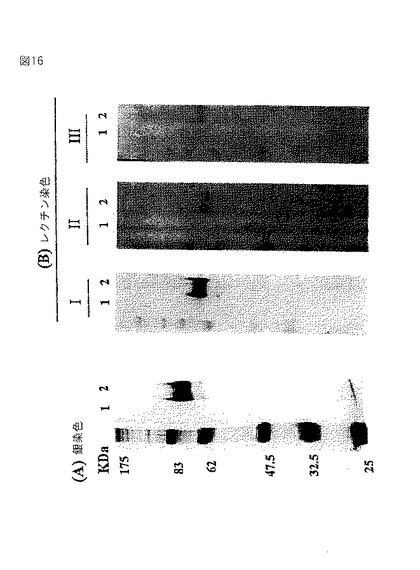
【図16】糖付加HRPの検出を示す、Ricinus communis 120 agglutinin(RCA120)アフィニティークロマトグラフィーの写真である。図16(A)は銀染色、そして図16(B)はレクチンRCA120での染色の結果である。レクチン染色のフィルターを細片に切断し、そして緩衝液単独(IおよびII)または過剰なガラクトースを含有する緩衝液(III)でプレインキュベートしたレクチンRCA120でプローブした。(II)では、HRPを、SDS−PAGEの前にDiplococcus pneumoniaeのβ−ガラクトシダーゼで処理した。レーン1はBY2−HRPからの収集画分;そしてレーン2はGT6−HRPからの収集画分である。左側の数字は、タンパク質の標準品の位置およびサイズ(KDa)を示す。
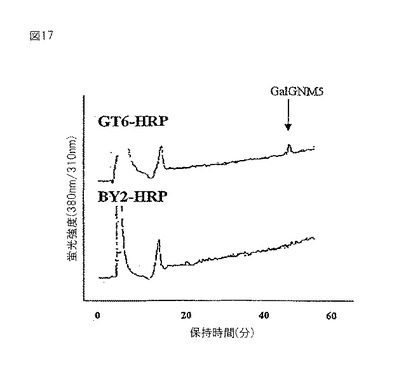
【図17】RCA120アフィニティークロマトグラフィーの後に精製されたHRPからのPA糖鎖の逆相HPLCの結果を示す図である。
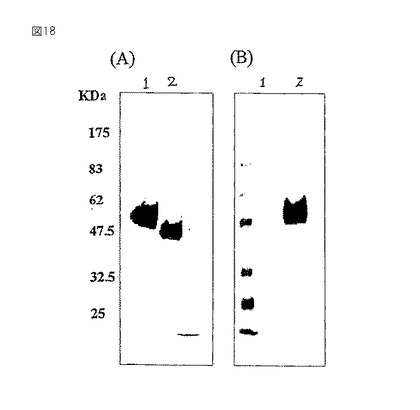
【図18】植物特異的な複合型グリカンの免疫検出を示すウェスタンブロッティングの写真である。精製されたHRPをSDS−PAGEで分画し、ニトロセルロースに転写し、そしてウサギ抗HRP(A)または植物の複合型グリカンに特異的な抗血清(B)でプローブした。レーン1=BY2−HRPからの精製HRP;レーン2=RCA120アフィニティークロマトグラフィー後のGT6−HRPからのガラクトシル化HRP。分子のサイズマーカーの位置を、KDaで左に示す。形質転換体GT6−HRP細胞由来のHRP上のガラクトシル化N−グリカンは、植物N−グリカンの指標であるβ1,2キシロース残基と特異的に反応することが示された抗血清と反応しなかった。
【図19】小胞体およびゴルジ体中の糖鎖修飾経路に関与するN−結合グリカンの構造および酵素。白抜き菱形はグルコースを、白抜き四角はGlcNAcを、白抜き丸はマンノースを、黒丸はガラクトースを、そして黒四角はシアル酸をそれぞれ示す。
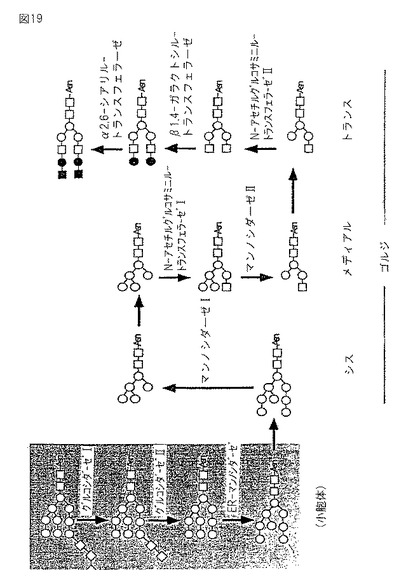
【図20】GT6細胞株中のN−結合グリカン類の構造、および各N−結合グリカンの比率を、同様に測定された野生型BY2細胞株それらとともに示す。白抜き四角はGlcNAcを、白抜き丸はマンノースを、黒丸はガラクトースを、そして黒四角はシアル酸をそれぞれ示す。
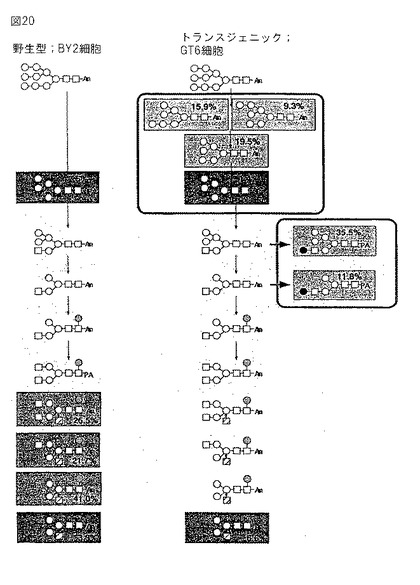
【図21】本発明の1つの実施態様を図示する。GT6細胞株において、アイソマー類Man7−、Man6−およびMan5GlcNAc2が認められた。これらの高マンノース型オリゴサッカライドは、いくつかのグリカンプロセッシング酵素によって、β1,4−ガラクトシルトランスフェラーゼ(Gal T)の基質であるように変換されるので、GlcNAc I、Man IおよびMan II cDNA類の導入は、オリゴサッカライドMan7−5GlcNAc2を、GalTの基質であり得るGlcNAcMan3GlcNAc2により効率的に導き得る。
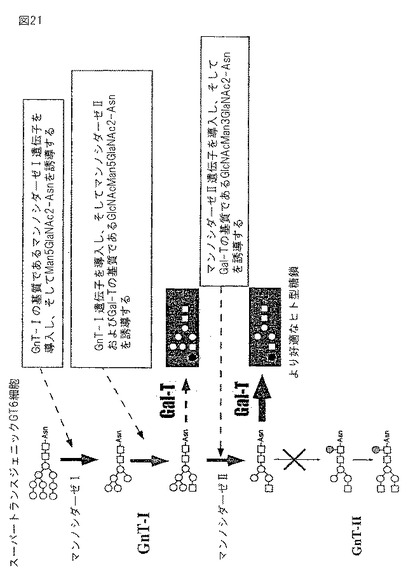
【図22】この図もまた、本発明の1つの実施態様を示す。1,4−ガラクトシルトランスフェラーゼ(Gal T)は、UDP−ガラクトースをドナー基質として用い、そしてGlcNAc2Man3GlcNAc2をアクセプター基質として用いる。UDP−ガラクトースのオリゴサッカライドを産生し得る。効率的な供給は、Gal T酵素反応を増大し、そしてより多くのガラクトシル化オリゴサッカライドを産生し得る。
【発明を実施するための形態】
【0019】
以下、本発明を詳細に説明する。
【0020】
本発明を実施することにおいて、他に特定されない限り、当該分野で公知である、タンパク質の分離および分析法、ならびに免疫学的手法が採用され得る。これらの手法は、市販のキット、抗体、標識物質などを使用して行い得る。
【0021】
本発明の方法は、ヒト型糖鎖をもつ糖タンパク質の生産方法に関する。本明細書において、「ヒト型糖鎖」とは、N−アセチルグルコサミン残基と結合したガラクトース残基を有する糖鎖をいう。ヒト型糖鎖におけるガラクトース残基は糖鎖の末端であってもよいし、ガラクトース残基のさらに外側にシアル酸残基が結合していてもよい。本発明の糖タンパク質は、ヒト型糖鎖のコア糖鎖部分、分岐糖鎖部分、および末端糖鎖部分からなる1つ以上の部分において、キシロースおよびフコースの少なくとも一方が結合していないことが好ましく、ヒト型糖鎖のいずれの部分においてもキシロースおよびフコースの少なくとも一方が結合していないことがより好ましく、最も好ましくはヒト型糖鎖中にキシロースおよびフコースのいずれも含まないことがさらにより好ましい。
【0022】
植物細胞とは、所望の任意の植物細胞であり得る。植物細胞は、培養細胞、培養組織中の細胞、培養器官中の細胞、または植物体中の細胞のいずれの形態であってもよい。好ましくは、植物細胞は、培養細胞、培養組織中の細胞、または培養器官中の細胞であり、最も好ましくは、この植物細胞は、ヒト型糖鎖をもつ糖タンパク質を産生する、完全な植物、またはその一部にある細胞である。本発明の生産方法に使用され得る植物種は、遺伝子導入を行い得る任意の植物種であり得る。本発明の生産方法に使用され得る植物種の例としては、ナス科、イネ科、アブラナ科、バラ科、マメ科、ウリ科、シソ科、ユリ科、アカザ科、セリ科の植物が挙げられる。
【0023】
ナス科の植物の例としては、Nicotiana、Solanum、Datura、Lycopersion、またはPetuniaに属する植物が挙げられ、例えば、タバコ、ナス、ジャガイモ、トマト、トウガラシ、ペチュニアなどを含む。
【0024】
イネ科の植物の例としては、Oryza、Hordenum、Secale、Scccharum、Echinochloa、またはZeaに属する植物が挙げられ、例えば、イネ、オオムギ、ライムギ、ヒエ、モロコシ、トウモロコシなどを含む。
【0025】
アブラナ科の植物の例としては、Raphanus、Brassica、Arabidopsis、Wasabia、またはCapsellaに属する植物が挙げられ、例えば、大根、アブラナ、シロイヌナズナ、ワサビ、ナズナなどを含む。
【0026】
バラ科の植物の例としては、Orunus、Malus、Pynus、Fragaria、またはRosaに属する植物が挙げられ、例えば、ウメ、モモ、リンゴ、ナシ、オランダイチゴ、バラなどを含む。
【0027】
マメ科の植物の例としては、Glycine、Vigna、Phaseolus、Pisum、Vicia、Arachis、Trifolium、Alphalfa、またはMedicagoに属する植物が挙げられ、例えば、ダイズ、アズキ、インゲンマメ、エンドウ、ソラマメ、ラッカセイ、クローバ、ウマゴヤシなどを含む。
【0028】
ウリ科の植物の例としては、Luffa、Cucurbita、またはCucumisに属する植物が挙げられ、例えば、ヘチマ、カボチャ、キュウリ、メロンなどを含む。
【0029】
シソ科の植物の例としては、Lavandula、Mentha、またはPerillaに属する植物が挙げられ、例えば、ラベンダー、ハッカ、シソなどを含む。
【0030】
ユリ科に属する植物の例としては、Allium、Lilium、またはTulipaに属する植物が挙げられ、例えば、ネギ、ニンニク、ユリ、チューリップなどを含む。
【0031】
アカザ科の植物の例としては、Spinaciaに属する植物が挙げられ、例えば、ホウレンソウを含む。
【0032】
セリ科の植物の例としては、Angelica、Daucus、Cryptotaenia、またはApitumに属する植物が挙げられ、例えば、シシウド、ニンジン、ミツバ、セロリなどを含む。
【0033】
本発明の生産方法に用いられる植物は、好ましくはタバコ、トマト、ジャガイモ、イネ、トウモロコシ、ダイコン、ダイズ、エンドウ、ウマゴヤシ、およびホウレンソウであり、より好ましくは、タバコ、トマト、ジャガイモ、トウモロコシ、およびダイズである。
【0034】
本明細書において、「非還元末端アセチルグルコサミン残基へのガラクトース残基の転移反応を行い得る酵素」とは、植物細胞内の糖タンパク質のタンパク質部分の合成後、糖鎖付加の際に生じる非還元末端アセチルグルコサミン残基へガラクトース残基を転移し得る酵素である。このような酵素の例としては、ガラクトシルトランスフェラーゼ、ラクトースシンターゼ、β−ガラクトシダーゼが挙げられる。このような酵素は、任意の動物種に由来し得るが、哺乳動物に由来することが好ましく、ヒトに由来することがより好ましい。
【0035】
本明細書において、「非還元末端アセチルグルコサミン残基へのガラクトース残基の転移反応を行い得る酵素の遺伝子」は、この酵素をコードすることが公知のヌクレオチド配列を用いて任意の動物細胞から単離してもよいし、市販のものを購入してもよいし、これらを植物での発現に適切なように改変して用いてもよい。
【0036】
本明細書では、「遺伝子」とは、構造遺伝子部分をいう。遺伝子には、植物での発現に適切なように、プロモーター、オペレーター、およびターミネーターなどの制御配列が連結され得る。
【0037】
本明細書において、「異種糖タンパク質」とは、本発明に用いられる植物において本来発現されない糖タンパク質をいう。異種糖タンパク質の例としては、酵素、ホルモン、サイトカイン、抗体、ワクチン、レセプター、血清タンパク質などが挙げられる。酵素の例としては、西洋ワサビペルオキシダーゼ、キナーゼ、グルコセレブロシダーゼ(glucocerebrosidase)、α−ガラクトシダーゼ、フィターゼ、TPA(tissue−type plasminogen activator)、HMG−CoAレダクターゼ(HMG−CoA reductase)などが挙げられる。ホルモンおよびサイトカインの例としては、エンケファリン、インターフェロンアルファ、GM−CSF、G−CSF、絨毛性性腺刺激ホルモン、インターロイキン−2、インターフェロン−ベータ、インターフェロン−ガンマ、エリスロポイエチン、血管内皮細胞増殖因子(vascular endothelial growth factor)、ヒト絨毛性ゴナドトロピン(HCG)、黄体形成ホルモン(LH)、甲状腺刺激ホルモン(TSH)、プロラクチン、卵胞刺激ホルモンなどが挙げられる。抗体の例としては、IgG、scFvなどが挙げられる。ワクチンの例としては、B型肝炎表面抗原、ロタウイルス抗原、大腸菌エンテロトキシン、マラリア抗原、狂犬病ウイルスrabies virusのGタンパク質、HIVウイルス糖タンパク質(例えば、gp120)などが挙げられる。レセプターおよびマトリックスタンパク質の例としては、EGFレセプター、フィブロネクチン、α1−アンチトリプシン、凝固因子VIIIなどが挙げられる。血清タンパク質の例としては、アルブミン、補体系タンパク質、プラスミノーゲン、コルチコステロイド結合グロブリン(corticosteroid−binding globulin)、スロキシン結合グロブリン(Throxine−binding globulin)、プロテインC(protein C)などが挙げられる。
【0038】
本明細書において、「異種糖タンパク質の遺伝子」は、目的の異種糖タンパク質をコードすることが公知のヌクレオチド配列を用いて任意の細胞から単離してもよいし、市販のものを購入してもよいし、これらを植物での発現に適切なように改変して用いてもよい。
【0039】
非還元末端アセチルグルコサミン残基へのガラクトース残基の転移反応を行い得る酵素および異種糖タンパク質の遺伝子は、当該分野で公知の方法により、植物細胞へ導入される。これらの遺伝子は、別々に導入してもよいし、同時に導入してもよい。植物細胞への遺伝子の導入方法の例としては、アグロバクテリウム法、エレクトロポレーション法、金粒子法などが挙げられる。
【0040】
遺伝子が導入された植物細胞は、当該分野で公知の方法により、導入された遺伝子の発現が確認され得る。このような確認方法としては、銀染色、ウェスタンブロッティング、ノザンハイブリダイゼーション、酵素活性の検出などが挙げられる。導入された遺伝子を発現する細胞は、形質転換細胞である。
【0041】
非還元末端アセチルグルコサミン残基へのガラクトース残基の転移反応を行い得る酵素および異種糖タンパク質を発現する形質転換細胞は、ヒト型の糖鎖を有する異種糖タンパク質を発現する。つまり、このようにして得られた形質転換植物は、ヒト型の糖鎖付加機構を有する。形質転換細胞を培養することにより、ヒト型の糖タンパク質が大量に生産され得る。ヒト型の糖タンパク質は、コア糖鎖および外部糖鎖を含み、このコア糖鎖は、複数のマンノースおよびアセチルグルコサミンから本質的になる。得られる糖タンパク質の外部糖鎖は、非還元末端糖鎖部分を含む。外部糖鎖は、直鎖状構造をもっていても分岐状構造をもっていてもよい。分岐糖鎖部分が、モノ、バイ、トリ、またはテトラ構造のいずれかであり得る。形質転換細胞により生産される糖タンパク質は、好ましくは、フコースまたはキシロースを含まない。
【0042】
得られた形質転換植物細胞は、培養細胞の状態で維持されてもよいし、特定の組織または器官へと分化させてもよいし、完全な植物体に再生させてもよい。あるいは、完全な植物体から得られる、種子、果実、葉、根、茎、花などの部分であってもよい。
【0043】
形質転換植物細胞により生産された、ヒト型の糖鎖をもつ糖タンパク質は、植物細胞から単離または抽出されてもよい。糖タンパク質の単離方法は、当該分野で公知である。あるいは、本発明の糖タンパク質は、形質転換細胞中に含まれたままの状態で食用に供され得る。本発明の糖タンパク質は、ヒト型の糖鎖付加を有するので、抗原性を有さず、それゆえ、ヒトを含む動物への投与に適している。
【0044】
以後、本発明を、制限ではない例示の実施例によって詳細に記載する。
【実施例】
【0045】
(実施例1)ヒトβ1−4ガラクトース転移酵素遺伝子のクローニング
β1−4ガラクトース転移酵素(hGT)(EC2.4.1.38)は、すでにクローン化されており、400アミノ酸からなる一次構造が明らかにされている(Masri,K.A.ら,Biochem.Biophys.Res.Commun.,157,657−663,1988)。
【0046】
(1)プライマーの作製と鋳型DNA
Masri.らの報告を参考にし、以下のようなプライマーを作製した。
hGT−5Eco:5’−AAAGAATTCGCGATGCCAGGCGCGCGTCCCT−3’(配列番号1)
hGT−2Sal:3’−TCGATCGCAAAACCATGTGCAGCTGATG−5’(配列番号2)
hGT−7Spe:3’−ACGGGACTCCTCAGGGGCGATGATCATAA−5’(配列番号3)
hGT6Spe:5’−AAGACTAGTGGGCCCCATGCTGATTGA−3’(配列番号4)
鋳型DNAは、Clontech社から購入したヒトゲノムDNA、ヒト胎盤cDNA、ヒト腎臓cDNAを用いた。
【0047】
(2)hGT遺伝子cDNAのクローニング
(i)ヒトゲノムDNAを鋳型とし、hGT−5EcoとhGT−7Speとをプライマーに用い;および(ii)ヒト胎盤cDNAを鋳型とし、hGT−2SalとhGT6Speとをプライマーに用いた、2つの組み合わせで以下の条件でPCR反応を行い、hGTをコードする領域を含む0.4kbおよび0.8kbの断片を得た。
(PCR反応系)鋳型DNA1μl、10×PCR緩衝液5μl、dNTPs(200μM)4μl、プライマー(10pmol)、Tagポリメラーゼ(宝酒造(株))0.5μl(Tubポリメラーゼの場合は0.2μl)に水を加えて50μlとした。
(PCR反応条件)第1段階:サイクル数1、変性(94℃)5分、アニーリング(55℃)1分、伸長(72℃)2分。第2段階:サイクル数30、変性(94℃)1分、アニーリング(55℃)1分、伸長(72℃)2分。第3段階:サイクル数1、変性(94℃)1分、アニーリング(55℃)2分、伸長(72℃)5分。
【0048】
得られた2つの断片を組み合わせてhGT遺伝子cDNAを構築し、pBluescriptIISK+(SK)にサブクローニングした。pBluescriptIISK+(SK)は、Stratagene社から購入した。図2に、hGT遺伝子cDNAを含むプラスミドの構築を示す。得られたhGT遺伝子の塩基配列を配列番号5に、そして推定されるアミノ酸配列を配列番号6に示す。
【0049】
得られた配列は、Masriら(上述)に開示のhGT配列と比較して以下の点で異なっていた。a)位置528のAがGに、位置562のCがTに、位置1047のAがGにそれぞれ変わっていたがコードされるアミノ酸に変化はなかった。b)位置622から630の9塩基が欠失していた。c)得られた上記の0.4kbおよび0.8kbの断片を接続するためにプライマー作製時に位置405のGをAに、位置408のTをAにそれぞれ変換した。なお、hGT遺伝子cDNAには2つの開始コドン(ATG)があるが、本実験では、第2番目の開始コドン(位置37)から翻訳が始まるように設計した。
【0050】
(実施例2)hGT遺伝子のタバコ培養細胞への導入
(1)hGTは大腸菌で活性型として発現することが報告されている(Aoki, D.ら,EMBO J.,9,3171,1990、およびNakazawa,K.ら,J.Biochem.,113,747,1993)。
【0051】
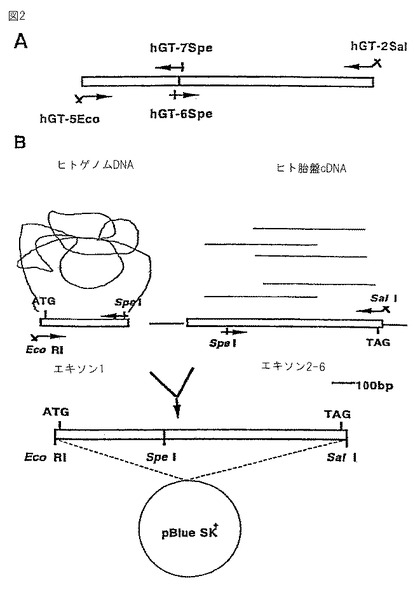
hGTをタバコ培養細胞で発現するために、発現用ベクターpGAhGTを図3に示すように構築した。プロモーターとしては、植物細胞内で構成的に発現するカリフラワーモザイクウイルス35Sプロモーター(CaMV 35Sプロモーター)を用いた。選択マーカーにはカナマイシン耐性遺伝子を用いた。pGAhGTをアグロバクテリウムを介してタバコ培養細胞に導入した。
【0052】
アグロバクテリウムの形質転換は、Bevan.ら,のtriparental mating法(Bevan,M.,Nucleic Acid Res.,12,8711,1984)を用いて行った。pGA系プラスミド(An,G.,Methods Enzymol.153,292,1987)をもつ大腸菌Escherichia coli DH5α株(suE44、ΔlacU169、(φ80lacZΔM15)、hsdR17)(Bethesda Research Laboratories Inc.:Focus 8(2)、9(1986))、およびヘルパープラスミドpRK2013(Bevan,M.,Nucleic Acid Res.,12,8711,1984)をもつ大腸菌Escherichia coli HB101を、それぞれ12.5mg/lのテトラサイクリン、50mg/lのカナマイシンを含む2×YT培地で37℃で1晩、アグロバクテリウムAgrobacterium tumefaciens EHA101株(Elizanbeth,E.H.,J.Bacteriol.,168,1291,1986)を、50mg/lのカナマイシン、25mg/lのクロラムフェニコールを含む2×YT培地で28℃で2晩培養した。各培養液1.5mlをエッペンドルフチューブにとり集菌した後、LB培地で3回洗浄した。得られた菌体をそれぞれ100μlの2×YT培地に懸濁した後、3種類の菌を混合し、2×YT寒天培地に塗抹し、28℃で培養してpGA系プラスミドを大腸菌からアグロバクテリウムに接合伝達させた。2日後、2×YT寒天培地上で一面に増殖した菌体の一部を白金耳でかきとり、50mg/lのカナマイシン、12.5mg/lのテトラサイクリン、25mg/lのクロラムフェニコールを含むLB寒天培地上に塗布した。28℃で2日間培養した後、単一コロニーを選択した。
【0053】
タバコ培養細胞の形質転換は、An,G.,Plant Mol.Bio.Mannual,A3,1.に記載の方法により行った。テトラサイクリン12.5mg/lを含むLB培地で28℃で36時間培養したアグロバクテリウム(pGA系のプラスミドをもつEHA101株)と培養4日目のタバコ培養細胞Nicotiana tabacum L.cv.bright yellow 2(理化学研究所ライフサイエンス筑波研究センター、ジーンバンク室植物細胞開発銀行のカタログ番号RPC1より細胞株名BY−2として入手した)の懸濁液をそれぞれ100μl、4mlずつシャーレに入れてよく混ぜ、25℃に暗所で静置した。2日後シャーレの中の培養液を遠心管に移して遠心分離(1000rpm、5分)により上澄みを除いた。次に新しい培地を入れて遠心分離し、細胞を150〜200mg/lのカナマイシン、250gmg/lのカルべニシリンの入った改変LS寒天培地のプレートに塗抹し、25℃暗黒下で静置した。約2〜3週間後にカルス化した細胞を新しいプレートに移植し、増殖しているクローンを選択した。さらに2〜3週間後に、カナマイシン、カルベニシリンを加えた改変LS培地30mlに移し、継代培養を行った。約1ケ月間選択を繰り返した。得られたいくつかの耐性株の中から無作為に6つの耐性株(GT1、4、5、6、8、および9)を選択した。
【0054】
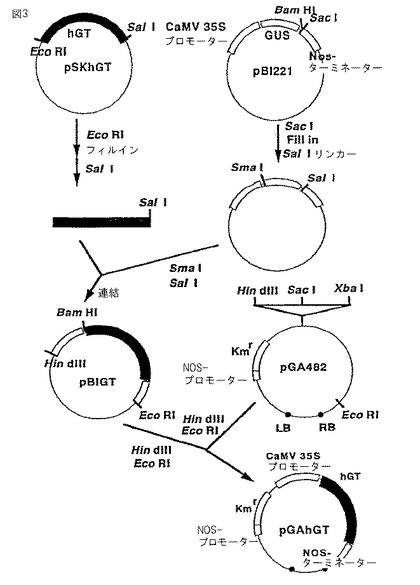
(2)導入されたhGT遺伝子の確認
得られた耐性株についてT−DNA中の、CaMV35Sプロモーター−hGT遺伝子cDNA−NOSターミネーターを含む2.2kbの断片がタバコ培養細胞のゲノムDNA中に組み込まれていることをサザン解析により確認した。上記の各耐性株からゲノムDNAを調製し、EcoRI、HindIIIで消化した後、サザン解析を行った。
【0055】
タバコ培養細胞からの染色体DNAの調製は、渡部の方法(渡部格、「クローニングとシーケンス」、植物バイオテクノロジー実験マニュアル、農村分化社)に従って行った。まず最初に、タバコ培養細胞10mlを液体窒素で凍結し、乳鉢、乳棒を用いて、粉末状になるまで粉砕した。得られた約5gの粉末が融解しないうちに、遠心管(40ml)中で60℃に予熱した5mlの2×CTAB(セチルトリメチルアンモニウムブロミド(cetyltrimethylammonium bromide))液に入れてゆっくりよく混ぜ、60℃で10分間以上、時々混ぜながら保温した。クロロホルム:イソアミルアルコール(24:1)5mlを加え、エマルジョンができるまでよく混ぜてから、遠心分離(2,800rpm、15分、室温)した。上層を新しい40ml遠心管に移し、クロロホルム:イソアミルアルコール(24:1)を用いて抽出操作を繰り返した。得られた上層に1/10容量の10%CTABを入れてよく混合した後、遠心分離操作を行った(2,800rpm、15分、室温)。上層を新しい遠心管に移し、1容量の冷イソプロパノールを入れて良く混ぜ、遠心分離(4,500rpm、20分、室温)した。上清液をアスピレータで除いた後、5mlの1M塩化ナトリウムを含むTE緩衝液を入れ、55〜60℃で完全に溶解した。これに、5mlの冷イソプロパノールを入れ、DNAが見えたら、チップの先に引っかけてエッペンドルフチューブ(80%冷エタノール入り)に移しリンスした。さらにDNAを70%エタノールでリンスし、乾燥した沈殿を、適当量のTE緩衝液に溶かし、5μlのRNAaseA(10mg/ml)を加え、37℃で1時間反応させた:2×CTAB液の組成;2%CTAB、0.1M Tris−HCl(pH8.0)、1.4M塩化ナトリウム、1%ポリビニルピロリドン(PVP);10%CTAB液の組成;10%CTAB、0.7M塩化ナトリウム。
【0056】
サザン解析は、以下のように行った。
【0057】
(i)DNAの電気泳動およびアルカリ変性:得られた染色体DNA40μgを制限酵素で完全分解した後、標準的な方法を用い、1.5%アガロースゲル電気泳動(50V)を行った。ゲルをエチジウムブロマイドで染色し、写真撮影した後、400mlの0.25M HCl中で20分間振とうした後、液を捨て、400mlの変性溶液(1.5M NaCl、0.5M NaOH)にゲルを浸し、45分間ゆっくり振とうした。次いで液を捨て、400mlの中和溶液(1.5M NaCl、0.5M Tris−Cl(pH7.4))を入れて15分間ゆっくり振とうした後液を捨て、再び中和溶液を400ml入れて15分間ゆっくり振とうした。(ii)トランスファー:電気泳動後のDNAを、20×SSCを用いてナイロンメンブレン(Hybond−N Amersham)にトランスファーした。トランスファーは12時間以上行った。プロットしたメンブレンを、室温で、1時間乾燥させた後、5分間UV固定を行った。20×SSCの組成:3M NaCl、0.3Mクエン酸ナトリウム。(iii)DNAプローブの調製:DNAプローブの調製は、Random prime Labeling Kit(宝酒造)を用いて行った。エッペンドルフチューブに次の反応液を調製し、95℃、3分間加熱後、氷中で急冷した;鋳型DNA 25ng、Random Primer 2μl、水を加えて5μlとする。10×緩衝液、dNTPをそれぞれ2.5μl、[α−32P]dCTP(1.85MBq、50mCi)を5μl加え、H2Oで24μlにフィルアップした。Klenow fragment 1μlを加え、37℃で10分間保温した後、NAP10カラム(Pharmacia社製)で溶出させてDNAを精製した。95℃で3分間加熱した後、氷中で急冷し、ハイブリダイゼーションブローブとした。(iv)ハイブリダイゼーション:0.05mg/ml 0.5%(w/v)SDSを、以下のプレハイブリダイゼーション溶液に加えて、上記(ii)のメンブレンを浸漬し、42℃で2時間以上プレハイブリダイゼーションを行った。その後、(iii)で調製したDNAプローブを加え、42℃で12時間以上ハイブリダイゼーションを行った:プレハイブリダイゼーション溶液の組成;5×SSC、50mMリン酸ナトリウム、50%(w/v)ホルムアミド、5×デンハルト溶液(100×デンハルト溶液を希釈して使用)、0.1%(w/v)SDS。100×デンハルト溶液の組成:2%(w/v)BSA、2%(w/v)Ficol 400、2%(w/v)ポリビニルピロリドン(PVP)。(v)オートラジオグラフィー:以下の順で洗浄した後、標準的な方法によりオートラジオグラフィーを行った。2×SSC、0.1%SDS中、65℃で15分間、2回、次いで0.1×SSC、0.1%SDS中、65℃で15分間、1回。
【0058】
上記で得られた耐性株のそれぞれから調製したゲノムDNAのサザン分析の結果を図4に示す。図4に示されるように、GT1、6、8および9の4株についてhGT遺伝子が組み込まれていることが確認された。
【0059】
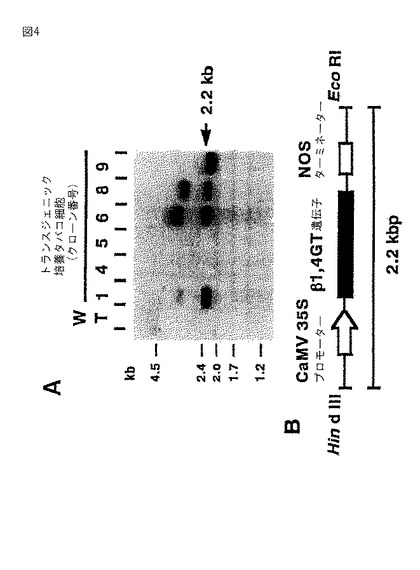
(実施例3)ガラクトシルトランスフェラーゼ形質転換体の解析
培養5〜7日目の形質転換体(GT−1、6、8、および9)および野生型のBY−2細胞の培養液からそれぞれ細胞を回収し、抽出緩衝液(25mM Tris−HCl、pH7.4;0.25Mスクロース、1mM MgCl2、50mM KCl)に懸濁し、そして超音波処理により破壊(200W;Kaijo Denki Co.,Japan)またはホモゲナイズした。細胞抽出液とミクロソーム画分を、Schwientek,T.らの方法(Schwientek,T.およびErnst,J.F.,Gene145,299−303,1994)に従って調製した。hGTタンパク質の発現は、抗ヒトガラクトシルトランスフェラーゼ(GT)モノクローナル抗体(MAb 8628;1:5000)(Uejima,T.ら,Cancer Res,52,6158−6163,1992;Uemura,M.ら,Cancer Res.,52,6153−6157,1992)(創価大学成松博士より供与)を用いてウェスタンブロッティングにより検出した。次いでブロットを西洋ワサビペルオキシダーゼ結合ヤギ抗マウスIgG(5%スキムミルク中1:1000;EY Laboratories,Inc.,CA)とインキュベートし、洗浄し、そして西洋ワサビペルオキシダーゼ発色反応は、POD免疫染色キット(Wako Chemicals,Osaka)を用いて実施した。
【0060】
植物特異的複合グリカンのイムノブロット分析は、ニンジン細胞壁β−フルクトシダーゼに対して惹起されたポリクローナル抗血清および西洋ワサビペルオキシダーゼ結合ヤギ抗ウサギIgG抗体(5%スキムミルク中1:1000;Sigma)を用いて実施した(Lauriere,M.ら,Plant Physiol.90,1182−1188,1989)。
【0061】
β1−4ガラクトシルトランスフェラーゼ活性は、基質として、UDP−ガラクトースおよびピリジルアミノ標識(PA−)GlcNAc2Man3GlcNAc2(GlcNAc2Man3GlcNAc2−PA)を用いてアッセイした(Morita,N.ら,J.Biochem.103,332−335,1988)。酵素反応液は、1〜120μgのタンパク質、25mMカコジル酸ナトリウム(pH7.4)、10mM MnCl2、200μM UDP−ガラクトース、および100nM GlcNAc2Man3GlcNAc2−PAを含んでいた。反応産物は、PALPAK Type RおよびType Nカラム(Takara社製)を製造業者の推奨する処方に従って用いてHPLCにより分析した。PA標識した標準として用いたGlcNAc2Man3GlcNAc2−PA、Gal2GlcNAc2Man3GlcNAc2−PAおよびGalGlcNAc2Man3GlcNAc2−PAの2つのアイソマーは、TAKARA SHUZOおよびHonen,Co.から購入した。
【0062】
図5に、形質転換体および野生型由来のタンパク質のイムノブロッティングを示す。図5に示すように、分子量約50kDaのポジティブシグナルが観察された。これは、アミノ酸配列から推定される分子量(約40kDa)より大きく、腹水から精製された、または酵母で発現されたウシガラクトシルトランスフェラーゼ(Uemura,M.ら,Cancer Res.52,6153−6157,1992;Schwientek,T.ら,J.Biol.Chem.271(7),3398−3405,1996)とほぼ等しかった。細胞溶解液(図5、レーン1〜4)と比較してミクロソーム画分(図5、レーン6〜8)で観察された強い免疫反応性バンドは、hGTが細胞内に優先的に局在化していたことを示す。野生型細胞には、免疫反応性のバンドは検出されなかった。
【0063】
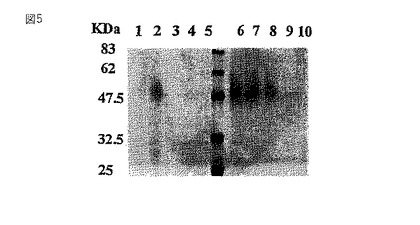
形質転換体GT6および野生型BY−2のミクロソーム画分のタンパク質を、RCA120アガロースカラム(Wako Chemicals,Osaka)に結合させ、15容量の10mM酢酸アンモニウムpH6.0で洗浄した。次いで0.2Mラクトースで結合したタンパク質を溶出し、SDS−PAGEで分画した後、銀染色(Wako Silver Staining Kit)(図6)またはレクチン染色した(図7)。レクチン染色には、膜ブロットをTTBS緩衝液(10mM Tris−HCl、pH7.4;0.15M NaCl、0.05%Tween20)中で洗浄し、西洋ワサビペルオキシダーゼ標識RCA120(Honen Co.社製)とインキュベートし、そしてPOD免疫染色キット(Wako Chemicals,Osaka)を用いてガラクトシル化されたグリカンを可視化した(図7)。図7に示すように、野生型のBY2細胞ではRCA120の結合は観察されなかったが、GT6は、グリカン成分の非還元末端にガラクトースを持つ糖タンパク質を有していた。
【0064】
野生型BY2細胞およびGT6細胞からのタンパク質抽出物およびRCA120アフィニティークロマトグラフィーからのGT6の溶出タンパク質を、複合グリカンに特異的なポリクローナル抗血清でプローブした(図8)。用いた抗血清は主に植物糖タンパク質上のβ1,2−キシロース残基に結合する(Lauriere,M.ら,Plant Physiol.90,1182−1188,1989)。図8に示すように、野生型BY2細胞(レーン1)は用いたポリクローナル抗血清と反応するいくつかの糖タンパク質を含んでいた。GT6は抗血清と反応する糖タンパク質の量が減少した(レーン2)。RCA120アフィニティークロマトグラフィーから溶出したGT6糖タンパク質は、ポリクローナル抗血清に結合せず、ガラクトシル化されたグリカンはβ1,2−キシロース残基を含んでいないことを示した(レーン3)。
【0065】
(実施例4)hGT導入タバコ培養細胞への西洋ワサビペルオキシダーゼ(HRP)遺伝子の導入
上記で得られたGT6株に西洋ワサビペルオキシダーゼを導入した。異なる型の植物ペルオキシダーゼの中で、西洋ワサビペルオキシダーゼ、特に中性アイソザイムCは、いわゆるHRP(EC1.11.1.7.)として、最もよく研究されている。HRPは、基質特異性が広く、安定性に優れることから種々の酵素反応、例えば、ウェスタンブロッティングの2次抗体などの酵素免疫法に利用されている。現在、西洋ワサビペルオキシダーゼアイソザイム遺伝子は多数クローニングされている(Fujiyama,K.ら,Eur.J.Biochem.,173,681−687,1988およびFujiyama,K.ら,Gene,89,163−169,1990)。それらのうちprxClaがコードするClaペルオキシダーゼ(ClaPRX)は、N末端側の30アミノ酸、C末端側の15アミノ酸からなるエキストラペプチドを含む353アミノ酸からなるタンパク質として翻訳され、その後プロセッシングをうけて、308アミノ酸からなる成熟型酵素となることが明らかにされた(Fujiyama,K.ら,Eur.J.Biochem.,173,681−687,1988)。ClaPRXの分子量は、42,200〜44,000であり、そのうち糖鎖が22〜27%を占めており、N−結合型糖鎖は8本存在することが知られている(Welinder,K.G.,Eur.J.Biochem.,96,483−502,1979)。ClaPRX遺伝子の導入は、図9に示すHRP発現用バイナリーベクターpBIHm−HRPを用いて行った。
【0066】
なお、このpBIHm−HRPは、以下のようにして調製した。まず、HRPcDNAを保持する植物用発現ベクター35S−prxC1a(KawaokaA.ら,J.Ferment.Bioeng.,78,49−53,1994)から1.9kbpのHindIII−SacI断片を切り出して調製した。このHindIII−SacI断片は、0.8kbpのCaMV35Sプロモーターに続く完全長の1.1kbpのprxC1acDNA断片を含む。1.9kbpのHindIII−SacI断片を、バイナリーベクターであるpBI101HmB(Akama K.ら,Plant Cell Rep.12,7−11,1992)のHindIII−SacI部位に挿入した。HPT遺伝子(ハイグロマイシン耐性遺伝子)の3’末端側のBamHI部位は破壊してある。
【0067】
GT6株はカナマイシン耐性であるので、選択マーカーとしてハイグロマイシン耐性遺伝子であるhpt遺伝子(Griz,L.およびDavies,J.、Gene,25,179−188,1983)を用いた。HRPによるGT6株の形質転換は、Rempel.D.H.およびNelson,L.M.の方法(Rempel,H.C.およびNelson,L.M.、Transgenic Res.4:199−207,1995)に記載の方法により行った。同様に、コントロールのHRP形質転換体を得るために、野生型BY2細胞にHRP形質転換体を導入して、BY2−HRP株を得た。hGTとHRPの二重形質転換体GT6−HRPは、通常の形質転換体と同様の期間で取得することができた。
【0068】
(実施例5)二重形質転換タバコ培養細胞におけるHRPの発現の確認
得られた二重形質転換体GT6−HRP、コントロールのBY2−HRP、野生型(WT)について、以下に示す方法で、HRP活性発現について調べた。表1に示すように、HRP遺伝子導入形質転換体は、野生株に比べて、約5倍のペルオキシダーゼ活性を示した。
【0069】
【表1】

【0070】
野生型にHRP遺伝子を導入して得られたクローンBY2−HRPは、hGTとHRPの二重形質転換体GT6−HRPと同程度のペルオキシダーゼ活性を示した。
【0071】
(ペルオキシダーゼ活性測定)
タバコ培養細胞をSolution Dの入ったエッペンドルフチューブに入れ、ホモジナイザー(HomogenizerS−203、池田理化社製)を用いて粉砕した。遠心分離(12,000rpm、20分間、4℃)により上清液を回収し、これを粗酵素液とした。1mlのSolution A、1mlのSolution Bおよび2mlのSolution Cを混合し、25℃で5分間保温した。これにSolution Dで適当に希釈した粗酵素液を加え、25℃で3分間反応させた。0.5mlの1N HClを加えることにより反応を停止し、480nmの吸光度を測定した。対照としては、酵素添加前に1N HClを添加した液を用いた:
Solution A;1mM ο−aminophenol
Solution B;4mM H2O2
Solution C;200mM リン酸ナトリウム緩衝液(pH7.0)
Solution D;10mM リン酸ナトリウム緩衝液(pH6.0)
【0072】
次に、このペルオキシダーゼ活性の上昇が、HRPの発現によるものであるか否かを確認するために、等電点電気泳動で分離後、ゲルの活性染色を行った。等電点電気泳動は、BIO−RAD社製のモデル111ミニIEFセルを用いて行った。PAGEゲルサポートフィルムの疎水面をガラスプレートに密着させた後、キャスティングトレーにのせた。調製したゲル用溶液をサポートフィルムとキャスティングトレーの間に流し込んで、蛍光灯の下45分間光重合させた。作製したゲルに試料を塗布し、蒸留水で湿らせた泳動槽にある両グラファイト電極にゲルが接するように置き、100V 15分、200V 15分、450V 60分の条件で電気泳動を行った。ゲル用溶液(ゲル1枚分)の組成は以下の通りである:蒸留水2.75ml、アクリルアミド(25%T、3%C)1.0ml、25%グリセロール1.0ml、バイオライト(40%、pH3〜10)0.25ml、10%過硫酸アンモニウム7.5μl、0.1%リボフラビン−5’−リン酸ナトリウム25μl、TEMED 1.5μl。
【0073】
ペルオキシダーゼの活性染色は、関根らの方法(関根ら,植物細胞工学、6、71−75、1994)に従って行った。図10に示すように、BY2−HRP株と、GT6−HRP株において、野生株には存在しない顕著なバンドがpI7.8の位置に検出された。抗HRP抗体を用いたウェスタン解析を行った結果、同じpI7.8に対応する位置にシグナルが検出され、hGTとHRPの二重形質転換体GT6−HRPにおいてHRPが発現していることが確認された。
【0074】
(実施例6)形質転換細胞GT6細胞中のN−結合型糖鎖の構造解析
(糖鎖構造の解析方法)
形質転換細胞GT6細胞のN−結合型糖鎖構造を、逆相HPLCとサイズ分画HPLCを組み合わせて、さらに二次元PA化糖鎖マッピング、エキソグリコシダーゼ消化、イオンスプレータンデムマススペクトロメトリー(IS−MS/MS)(Perkin Elmer社製)により解析した。まず、細胞抽出液をアセトンで脱脂後、100℃、12時間ヒドラジン処理し、糖鎖部分を遊離させた。ヒドラジン分解物を、N−アセチル化し、Dowex 50X2およびDowex 1X2(それぞれ、代理店室町化学工業株式会社、The Dow Chemical Co.社製)により脱塩し、0.1NアンモニアによりSephadex G−25ゲルろ過カラム(1.8×180cm)(Pharmacia社製)を用いて分画した。ピリジルアミノ化(PA化)は先に述べた通りである。PA化糖鎖を、Jasco 821−FP Intelligent Spectrophotometer付きJasco880−PU HPLC装置(日本分光社製)により、Cosmosil 5C18−P及びAsahipak NH2P−50をカラムに用いて分離した。溶出位置を、自ら調製したか、あるいは購入した(Wako ChemicalsおよびTAKARA SHUZO)既知の標準品と比較した。
【0075】
N−アセチル−β−D−グルコサミニダーゼ(Diplococcus pneumoniae,Boehringer Mannheim)あるいはマンノシダーゼ(タチナタマメ、Sigma)を用いたグリコシダーゼ消化は、Kimura,Y.ら、Biosci.Biotech.Biochem.56(2)、215−222、1992で述べた方法と同様の条件下で約1nmolのPA化糖鎖について行った。β−ガラクトシダーゼ(Diplococcus pneumoniae,Boehringer Mannheim)とAspergillus saitoi由来のα−1,2マンノシダーゼ(東北大学吉田孝博士より分与)による消化については、50mM 酢酸ナトリウム緩衝液(pH5.5)中に1nmolのPA化糖鎖を200mU β−ガラクトシダーゼあるいは60μg α−1,2マンノシダーゼを37℃で加温した。酵素反応を沸騰させて停止し、消化物の一部をサイズ分画HPLCにより解析した。生じた消化物の分子量はイオンスプレーマススペクトロメトリー(IS−MS/MS)により解析し、そして/あるいはPalacpac,N.Q.ら、Biosci.Biotech.Biochem.63(1)35−39、1999およびKimuraら、Biosci.Biotech.Biochem.56(2)、215−222、1992に示されているように標準糖鎖と比較した。
【0076】
IS−MS/MS実験は、Perkin Elmer Sciex API−IIIにより行った。ポジティブモードで、イオンスプレーの電圧を4200Vにて行った。スキャンニングを0.5Da毎で行い、m/zを200から記録した。
【0077】
(GT6細胞の糖鎖の解析)
GT6細胞より調製したPA化糖鎖を精製し、逆相HPLCとサイズ分画HPLCを組み合わせて解析した。サイズ分画HPLCのもとで、10〜20分の位置のフラクションI(図11)には、N−結合型糖鎖は溶出しておらず、ヒドラジン分解による副産物を含む非吸着部分であることを示唆している。MS/MS解析では、PA−GlcNAcに相当する300というm/z値を持つフラグメントイオンを検出できなかった。同様に、50〜60分の位置のフラクションXIも、サイズ分画HPLCで溶出を示すピークを検出できなかったので、N−結合型糖鎖を含んでいないことがわかった。しかし、サイズ分画HPLC(図11)におけるフラクションIIからXまでの解析から、図12に示すA〜Qを含む全部で17のピークを回収および精製した。
【0078】
IS−MS/MS分析は、これらのピークのうち7つのピークがN−結合型糖鎖であることを示した。以下各ピークの分析結果を示す。
【0079】
オリゴ糖−A、−E、−H、−I、−M、−O、−P、及び−Q(図12)の溶出位置及び分子量は、PA化糖鎖標準品と対応せず、MS/MS解析ではPA−GlcNAc及びPA−GlcNAc2に相当する300及び503というm/z値を有していたが、N−結合型糖鎖一般的に認められるManGlcNA2(M1)或いはトリマンノースコア糖鎖Man3GlcNAc2(M3)に相当するフラグメントイオンを検出できなかった(データ未掲載)。他のピークのオリゴ糖−B,−D及び−Nも、300というm/z値を持つフラグメントイオンを検出できなかったので、N−結合型ではなかった。そこで残りの7つのN−結合型糖鎖について調べた。
【0080】
ピーク−C(m/z1637.5;モル存在比9.3%)、ピーク−F([M+2H]2+ m/z819.5,[M+H]+ m/z1639;モル存在比15.9%)、およびピーク−G(m/z1475.5 モル存在比19.5%)の溶出位置と分子量はそれぞれ、高マンノース型糖鎖Man7GlcNAC2(アイソマーM7A及びM7B)、およびMan6GlcNAc2(M6B)であった。タチナタマメ α−マンノシダーゼで消化すると、各N−結合型糖鎖は、サイズ分画HPLCによりManGlcNAc(M1)へと分解されていることを示した(データ未掲載)。消化物のIS−MS実験によりm/z値665.5というイオンは、M1の計算値664.66に対応していたことから、これらは対応する個々のPA化糖鎖標準品と構造的に同一であることを示している。
【0081】
ピーク−J(6.6%)は、分子量1121.5を有していたが、これはMan3Xyl1GlcNAc2−PA(M3X)の分子量計算値m/z1121.05と同じであった。989.5、827.5、665.5、503.3及び300に見られたフラグメントイオンの位置は、Man3Xyl1GlcNAc2−PAから逐次Xyl、Man、Man、Man及びGlcNAcがこの順序で遊離したことと矛盾しない。タチナタマメα−マンノシダーゼで処理すると、非還元末端のマンノース残基を除去でき、2次元マッピングはMan1Xyl1GlcNAc2−PAの溶出位置と同じであった(データ未掲載)。
【0082】
ピーク−K(13.2%)のIS−MS実験の解析結果は、このフラクションが2種のN−結合型糖鎖を含むことを示し、そのひとつは分子量が1314.0(1.4%)で他方が1354.5(11.8%)だった。このフラクションをさらに逆相HPLCに供し、精製し、更に解析した。分子量1314.0を有する糖鎖ピーク−K−1は、糖鎖標準品Man5GlcNAc2−PA(M5)と二次元マッピング及びm/z測定値が同一だった。さらに、タチナタマメα−マンノシダーゼで処理すると、その分解産物の溶出位置が2次元マッピングでM1と同じ位置に移動したことら、4つのマンノース残基が除去されたことを示した。
【0083】
(GT6細胞中のガラクトースが付加したN−結合型糖鎖)
m/z1354.5を有する糖鎖ピーク−K−2は、Gal1GlcNAc1Man3GlcNAc2−PA(GalGNM3)の予想される分子量m/z1354.3とよく一致していた。質量分析の結果から、親分子から派生したと考えられるフラグメントイオンはそれぞれ、m/z1193.5がGlcNAc1Man3GlcNAc2−PA、m/z989.5がMan3GlcNAc2−PA、m/z827.5がMan2GlcNAc2−PA、m/z665がManGlcNAc2−PA、m/z503がGlcNAc2−PA、m/z336がManGlcNAc、m/z300がGlcNAc−PAそしてm/z204がGlcNAcであった。推定されるN−結合型糖鎖構造から、GalGNM3の2種のアイソマーが考えられる(図13)。すなわち、Galβ4GlcNAcβ2Manα6(Manα3)Manβ4GlcNacβ4GlcNAc−PA及びManα6(Galβ4GlcNAcβ2Manα3)Manβ4GlcNAcβ4GlcNAc−PAである。精製したPA化糖鎖は、糖鎖標準品Manα6(Galβ4GlcNAcβ2Manα3)Manβ4GlcNAcβ4GlcNAc−PAと同一の逆相HPLCの溶出位置であった(図13のB)。
【0084】
糖鎖をエキソグリコシダーゼで処理し、そして糖鎖構造を確認した。D.pneumoniae β−ガラクトシダーゼはGalβ1−4GlcNAc結合特異的であるが、本酵素による消化物はGlcNAc1Man3GlcNAc2−PAとサイズ分画HPLCにて同一の位置に溶出した(図14A−II)。また、IS−MS/MS分析からm/z1192.0が得られた。これらの結果は、1つのガラクトース残基が、β1−4結合している非還元末端のGlcNAcから除去されたことを示している。さらにこの産物を、β1−2GlcNAc結合特異的なDiplococcus pneumoniae由来のN−アセチル−β−D−グルコサミニダーゼ(Yamashita,K.ら J.Biochem.93,135−147、1983)で消化すると、その消化物は標準品Man3GlcNAc2−PAとサイズ分画HPLCにて同じ位置に溶出した(図14A−III)。続いてタチナタマメα−マンノシダーゼで処理すると、標準品ManGlcNAc2−PAとサイズ分画HPLCにて同じ位置に溶出した(図14A−IV)。糖鎖構造を図15のK−2に示す。
【0085】
ピーク−L(35.5%)は、質量分析結果から[M+2H]2+が840、[M+H]+が1680.0であり、Gal1GlcNAc1Man5GlcNAc2−PA(GalGNM5)の予想される分子量m/z1678.55とよく一致していた。質量分析の結果から、親分子から派生したと考えられるフラグメントイオンはそれぞれ、m/z1313.5がMan5GlcNAc2−PA、m/z1152がMan4GlcNAc2−PA、m/z989.5がMan3GlcNAc2−PA、m/z827.5がMan2GlcNAc2−PA、m/z665がManGlcNAc2−PA、m/z503がGlcNAc2−PA、m/z336がManGlcNAc、m/z300がGlcNAc−PAそしてm/z204がGlcNAcであった。D. pneumoniae β−ガラクトシダーゼによる消化物はGlcNAc1Man5GlcNAc2−PAとサイズ分画HPLCにて同一の位置に溶出した(図14B−II)。この結果は、1つのガラクトース残基が、β1−4結合している非還元末端のGlcNAcに結合していたことを示している。ガラクトースが除去されたことを、IS−MS/MS分析により得られた分子量が[M+2H]2+が759、[M+H]+が1518.0であることにより確認した。親シグナルm/z1518.0であるGlcNAc1Man5GlcNAc2−PAから、フラグメントイオンm/z1314がMan5GlcNAc2−PA、m/z1152がMan4GlcNAc2−PA、m/z990がMan3GlcNAc2−PA、m/z827.5がMan2GlcNAc2−PA、m/z665.5がMan1GlcNAc2−PA、m/z503がGlcNAc2−PA、m/z300がGlcNAc−PAとが派生していた。GlcNAc1Man5GlcNAc2−PAをDiplococcus pneumoniae由来のN−アセチル−β−D−グルコサミニダーゼで消化すると、その消化物は標準品Man5GlcNAc2−PAとサイズ分画HPLCにて同じ位置に溶出した(図14B−III)。Aspergillus saitoi由来のα−1,2マンノシダーゼで処理しても溶出位置が移動しなかった(図14B−IV)。しかし、続いてタチナタマメ α−マンノシダーゼで処理すると、標準品Man1GlcNAc2−PAとサイズ分画HPLCにて同じ位置に溶出した(図14B−V)。このことは、非還元末端に存在する4つのマンノース残基が除去されたことを示している。これらの結果から、PA化糖鎖は、5つのいずれのマンノース残基も、α1−3結合したマンノース残基にα1−2結合していないことを示す。以上エキソグリコシダーゼ消化、2次元糖鎖マッピング、IS−MS/MS分析から導きだされる糖鎖構造は、GalGNM5で、図15のLに示す。
【0086】
図20は、GT6細胞株におけるN−結合グリカンの構造と各N−結合グリカンの比率に関する上記の結果を、同様に決定された野生型BY2細胞株におけるそれらとともに要約する。図20において、白抜き四角はGlcNAcを、白抜き丸はマンノースを、黒丸はガラクトースを、斜線をひいた四角はキシロースを、そして点をつけた丸はフコースをそれぞれ示す。
【0087】
GT6細胞株では、アイソマー類であるMan7−、Man6−およびMan5GlcNAc2が観察された。これらの高マンノース型オリゴサッカライドは、β1,4−ガラクトシルトランスフェラーゼ(GalT)の基質であるので、GlcNAcI、ManIおよびManII cDNAの導入は、オリゴサッカライドMan7−5GlcNAc2を、GalTの基質であり得るGlcNAcMan3GlcNAc2へより効率的に導き得る(図21)。
【0088】
GnT Iを欠くA.thaliana cglI変異体は、複合型N−グリカンを合成することができない(von Schaewen,A.,Sturm,A.,O’Neill、J.,およびChrispeels、MJ.,Plant Physiol.,1993 Aug;102(4):1109−1118、N−アセチルグルコサミニルトランスフェラーゼIを欠き、そしてゴルジ体で修飾される複合体N−結合グリカンを合成できない変異体Arabidopsis植物の単離)。このcglI変異体におけるヒトGnTでの相補性は、哺乳動物酵素が、植物N−グリコシル化経路に寄与し得たことを示した(Gomez、L.およびChrispeels,M.J.,Proc.Natl.Acad.Sci.USA 1994 3月 1;91(5)1829−1833、複合アスパラギン結合グリカンを欠くArabidopsis thalianaの、N−アセチルグルコサミニルトランスフェラーゼIをコードするヒトcDNAでの相補性)。さらに、A.thalianaから単離されたGnT I cDNAは、CHO Lec1細胞のN−アセチルグルコサミニルトランスフェラーゼI欠損を相補した(Bakker,H.,Lommen,A.,Jordi,W.,Stiekema,W.,およびBosch,D.,Biochem.Biophys.Res.Commun.,1999 8月 11;261(3):829−32、A.thaliana cDNAは、CHO Lec1細胞のN−アセチルグルコサミニルトランスフェラーゼI欠損を相補する)。ヒトManIおよびManIIをコードするcDNAが単離され、そして配列決定された(Bause,E.,Bieberich,E.,Rolfs,A.,Volker,C.およびSchmidt,B.,Eur J Biochem 1993 10月 15;217(2):535−40、ヒト腎臓からのMan9−マンノシダーゼの分子クローニングおよび一次構造;Tremblay,L.O.,Campbell,Dyke,N.およびHerscovics,A.,Glycobiology 1988 6月;8(6):585−95、N−グリカン成熟に関与する新規ヒトα1,2−マンノシダーゼ遺伝子の分子クローニング、染色体マッピングおよび組織特異的発現;およびMisago.M.,Liao,Y.F.,Kudo,S.,Eto,S.,Mattei,M.G.,Moremen,K.W.,Fukuda,M.N.,ヒトα−マンノシダーゼIIおよび従前には認識されていないα−マンノシダーゼIIxアイソザイムをコードするcDNAの分子クローニングおよび発現)。ヒトManIは、2つのアイソザイムManIAおよびManIBをもち、そしてアイソザイムのcDNAのヌクレオチド構造が示された(Bause,E.,ら、およびTremblay,L.O.,前述)。これらのcDNAを、BY細胞株中に形質転換することにより、ヒト型糖タンパク質を産生する効率的な細胞株が得られ得る。β1,4−ガラクトシルトランスフェラーゼ(GalT)は、UDP−ガラクトースをドナー基質として用い、そしてGlcNAc2Man3GlcNAc2をアクセプター基質として用いる。UDP−ガラクトースの効率的な供給はGalT酵素反応を増大し、そしてより多くのガラクトシル化オリゴサッカライドが産生される(図22)。
【0089】
(実施例7)二重形質転換体GT6−HRP細胞中のHRPの糖鎖の構造解析
GT6−HRP細胞またはコントロールのBY2−HRP細胞の7日間培養した培養液のそれぞれのホモゲナイズ物50gから得た粗細胞溶解液を、10mMリン酸ナトリウム緩衝液(pH6.0)で平衡化したCM Sepharose FFカラム(1×10cm)(Pharmacia社製)にかけた。カラムを洗浄後、溶出したペルオキシダーゼを403nmの吸光度で測定した。プールした画分を限外濾過(分子量カット:10,000、Advantec社製)により濃縮し、50mMのリン酸ナトリウム緩衝液(pH7.0)中で透析し、そして平衡化したベンズヒドロキサミン酸−アガロースアフィニティーカラム(1×10cm)(KemEn Tec,Denmark)にかけた。カラムを15倍容量の50mMリン酸ナトリウム緩衝液(pH7.0)で洗浄した後、同じ緩衝液中に調製した0.5Mのホウ酸で、結合したHRPを溶出した。得られたペルオキシダーゼ活性画分をプールし、透析および濃縮した。
【0090】
形質転換細胞GT6−HRP細胞およびBY2−HRP細胞のそれぞれから得た精製HRPを、1×10cmのRCA120−アガロースカラムにかけた。次いでカラムを、15倍容量の10mM酢酸アンモニウム(pH6.0)で洗浄した。結合タンパク質を、常法に従って溶出し、アッセイした。
【0091】
β1−4結合ガラクトースに特異的であるRCA120アフィニティークロマトグラフィーから溶出された精製HRPのレクチン染色では、レクチンRCA120が、形質転換細胞GT6−HRPで産生されたHRPのみを結合した。レクチンを競合するガラクトースとプレインキュベートすることによりレクチン結合は劇的に減少したので(図16bIII)、この結合は炭水化物特異的であった。D.pneumoniae β−ガラクトシダーゼで精製HRPを前処理しても、RCA120結合を阻害した。これらの結果は、RCAがHRP上のN結合型オリゴ糖鎖の末端β1−4結合位置にあるガラクトースに特異的に結合したことを示す。BY2−HRP細胞中にRCA結合糖タンパク質がないことは、この細胞がHRPグリカンの非還元末端にβ1−4結合ガラクトース残基を転移し得ないことを示す。
【0092】
RCA120を用いて精製したHRP由来のPA誘導体の逆相HPLC(Jasco821−FP Intelligent Spectrofluorometerを備えたJasco880−PU HPLC装置上、Cosmosil 5C18−PカラムまたはAsahipakNH2Pカラムを用いた)は、GT6−HRPより精製されたHRPタンパク質上の糖鎖が単一のピークを示した(図17)。一方、BY2−HRPより精製されたHRP画分からは、結合タンパク質または検出可能なピークは観察されなかった。GT6−HRPから得られたピークは、サイズ分画HPLC上で均一であった。二次元マッピング分析および標準糖鎖との同時クロマトグラフィーは、このオリゴサッカライドがGal1GlcNAc1Man5GlcNAc2−PAと一致することが判明した。この構造の確認は、連続的エクソグリコシダーゼ消化によりなされた。標準糖鎖としては、先に調製された糖鎖(Kimura,Yら、Biosci.Biotech.Biochem.56(2),215−222,1992)または市販(Wako Chemicals,OsakaおよびTAKARA SHUZO)の糖鎖を用いた。
【0093】
PA糖鎖のβ−ガラクトシダーゼ(D.pneumoniae)消化は、標準のGlcNAc1Man5GlcNAc2−PAの溶出位置と一致し、1つの非還元末端GlcNAcにβ1−4結合した1つのガラクトース残基が除去されたことを示した。D.pneumoniae由来のN−アセチル−β−D−グルコサミニダーゼでのこの消化物のさらなる消化は、Man5GlcNAc2−PAの溶出位置に等しい糖鎖を生じ、1つの非還元末端マンノース残基にβ1−2結合したGlcNAc残基を損失したことを示した。植物におけるN結合型プロセッシング経路に基づけば、除去されたGlcNAc残基は、β1−4マンノース残基に連結したα1−3マンノースにあると考えられる。GlcNAc残基の結合位置を確立するために、Man5GlcNAc2−PA(M5)を、Aspergillus saitoi由来α1−2マンノシダーゼとともにインキュベートした。予想されたように、溶出位置にシフトは見られず、M5が予想された構造Manα1−6(Manα1−3)Manα1−6(Manα1−3)Manβ1−4GlcNAcβ1−4GlcNAcを有することが確認された。糖鎖をタチナタマメα−マンノシダーゼを用いてさらに切りそろえると、公知のMan1GlcNAc2−PAの溶出位置になった。従って、この糖鎖構造は、Manα1−6(Manα1−3)Manα1−6(Galβ1−4GlcNAcβ1−2Manα1−3)Manβ1−4GlcNAcβ1−4GlcNAc(Gal1GlcNAc1Man5GlcNAc2)に対応する。以上の結果から、GT6細胞中に存在する糖鎖構造としては図15に示される通りであり、二重形質転換体GT6−HRP細胞由来のHRPタンパク質の有する糖鎖構造は、Manα1−6(Manα1−3)Manα1−6(Galβ1−4GlcNAcβ1−2Manα1−3)Manβ1−4GlcNAcβ1−4GlcNAc(Gal1GlcNAc1Man5GlcNAc2)であることが導き出される。
【0094】
同様に、形質転換細胞GT6−HRP由来のHRPのガラクトシル化N−グリカンは、植物のN−グリカンに特徴的なβ1,2キシロース残基と特異的に反応することが示されている抗血清と反応せず、植物の複合型グリカン中の抗原性を引き起こすことが示されている糖残基(キシロース残基)(Garcia−Casado,G.ら,Glycobiology 6(4):471−477,1996)の1つが存在しないことを示した(図18)。
【0095】
(発明の効果)
本発明により、ヒト型糖鎖をもつ糖タンパク質の生産方法;非還元末端アセチルグルコサミン残基へのガラクトース残基の転移反応を行い得る糖鎖付加機構を備え、この糖鎖付加機構が、コア糖鎖および外部糖鎖を含む糖鎖であって、コア糖鎖が複数のマンノースおよびアセチルグルコサミンから本質的になり、外部糖鎖が非還元末端ガラクトースを含む末端糖鎖部分を含む糖鎖、を付加する、植物細胞;およびこの方法によって得られたヒト型の糖鎖をもつ糖タンパク質が提供される。本発明のヒト型の糖鎖をもつ糖タンパク質は、糖付加がヒト型であるために、抗原性を有さない。それゆえ、ヒトを含む動物への投与のために有用である。
【技術分野】
【0001】
本発明は、植物による異種糖タンパク質の発現に関する。
【背景技術】
【0002】
生体内の機能的タンパク質の多くは糖鎖を持つ糖タンパク質である。糖タンパク質内の糖鎖の多様性は、いくつかの重要な役割を生理学的に担っていることが明らかにされている(Lain,R.A.,Glycobiology,4,759−767,1994)。
【0003】
最近、糖鎖の作用が2つのカテゴリーに分けられ得ることが明確になった。1つは、糖鎖が、血液からの糖タンパク質のクリアランス、リソソーム酵素のリソソームへのターゲッティング、糖タンパク質による特定の組織および器官へのターゲッティングにおいて、細胞間接着のリガンドとして、および細菌およびウイルスの受容体として直接的な機能を有する場合である。例えば、エイズウイルス(HIV)が標的細胞に感染する過程で、糖タンパク質の糖鎖が関与していることが示されている(Rabehi,L.ら、Glycoconj.J.,12,7−16,1995)。HIV表面は、エンベロープタンパク質gp120で覆われ、gp120の糖鎖が標的細胞のCD4に結合することでHIVウイルスの感染が始まる。糖鎖のもう1つの働きは、糖鎖自体は機能的分子ではないが、タンパク質の高次構造の形成、タンパク質の溶解性、タンパク質のプロテアーゼ抵抗性、抗原性の抑制、タンパク質機能の修飾、タンパク質代謝速度の調節および細胞膜における発現量の調節などに間接的に関与する場合である。例えば、神経系に広く分布する神経細胞接着分子の接着性は糖鎖により調節されている(Edelman,G.M.,Ann.Rev.Biochem.,54,135−169,1985)。
【0004】
真核生物では、糖タンパク質の糖鎖は、小胞体の脂質上で前駆体型糖鎖として合成される。糖鎖部分はタンパク質に転移され、次いでこのタンパク質上のいくつかの糖残基が除去され、ゴルジ体に輸送される。ゴルジ体において、過剰の糖残基が除去された後、さらなる糖残基(例えばマンノース)が付加され、そして糖鎖が伸長される(Narimatsu,H.,Microbiol.Immunol.,38,489−504,1994)。
【0005】
より詳細に述べれば、例えば、ドリコールアンカー(dolichol anchors)上のGlc3Man9GlcNAc2は、小胞体(ER)膜中のタンパク質に転移される(Moremen K.W.,Trimble、R.B.およびHerscovics A.,Glycobiology 1994 4月;4(2):113−25、アスパラギン連結オリゴサッカライドプロセッシング経路のグリコシダーゼ;およびSturm,A.1995 植物タンパク質のN−グリコシル化、New Comprehensive Biochemistry.Glycoproteins,第29a巻、Montreuil、J.,Schachter、H.およびVliegenthart、J.F.G.(編).Elsevier Science Publishers B.V.,The Netherland、521−541頁)。小胞体グリコシダーゼIおよびIIは、3つのグルコース単位を除去する(Sturm、A.1995、前述;およびKaushal G.P.およびElbein A.D.,1989、植物における糖タンパク質プロセッシング酵素。Methods Enzymology 179、Complex Carbohydrates Part F.Ginsburg V.(編)、Academic Press,Inc.NY、452−475頁)。得られる高マンノース構造(Man9GlcNAc2)は、小胞体マンノシダーゼによりトリミングされる(Moremen K.W.ら、前述;およびKornfeld,R.およびKornfeld,S.,Annu.Rev.Biochem.54、631−664、1985;アスパラギン連結オリゴサッカライドのアセンブリ)。除去されるマンノース残基の数は、プロセッシング酵素への接近可能性における差異に従って変動する。アイソマー類であるMan8−、Man7−、Man6−およびMan5GlcNAc2が、小胞体マンノシダーゼおよびマンノシダーゼIによるプロセッングの間に生産される(Kornfeld,R.およびKornfeld,S.,前述)。4つのマンノース残基がマンノシダーゼI(Man I)によって完全に除去されるとき、産物は、Man5GlcNAc2である。N−アセチルグルコサミニルトランスフェラーゼI(GlcNAcI)は、N−アセチルグルコサミン(GlcNAc)を、UDP−GlcNAcからMan5GlcNAc2へ転移し、GlcNAcMan5GlcNAc2を生じる(Schachter,H.,Narasimhan,S.,Gleeson,P.,およびVella,G.,グリコシルトランスフェラーゼは、複合型またはN−アセチルガラクトサミン型のN−グリコシル的に連結されたオリゴサッカライドの伸長に関与する。Methods Enzymol 98:Biomembranes Part L.Fleischer,S.,およびFleischer,B.(編)、Academic Press,Inc.NY.98−134頁、1983)。マンノシダーゼII(Man II)は、GlcNAcMan5GlcNAc2から2つのマンノース残基を除去し、GlcNAcMan3GlcNAc2を生じる(Kaushal,G.P.およびElbein,A.D.,前述;およびKornfeld,R.およびKornfeld,S.,前述)。オリゴサッカライドGlcNAcMan4GlcNAc2は、N−アセチルグルコサミニルトランスフェラーゼII(GlcNAcII)の基質として用いられる(Moremen,K.W.ら、前述;Kaushal,G.P.およびElbein,A.D.,前述;およびKornfeld,R.およびKornfeld,S.,前述)。図19は、N−結合グリカンの上記の構造、ならびに小胞体およびゴルジ体中の糖鎖修飾経路に関与する酵素を要約する。図19において、白抜きの菱形はグルコースを、白抜きの四角はGlcNAcを、白抜きの丸はマンノースを、黒丸はガラクトースを、そして黒四角はシアル酸をそれぞれ示す。
【0006】
このゴルジ体における糖付加は末端部糖鎖合成と呼ばれ、このプロセスは、生物種により大きく異なる。糖鎖生合成系は、真核生物の種によって異なり、その結果糖鎖構造は種特異的であり、そして糖を付加する糖転移酵素およびゴルジ体の進化を反映している(成松久、細胞工学、15,802−810,1996)。
【0007】
アスパラギン結合型(N−結合型)糖鎖に注目すると、動物には、高マンノース型糖鎖、複合型糖鎖およびそのハイブリッド型が存在する。図1にそれらの構造を示す。一方、植物の複合型糖鎖には、動物の糖鎖にはないα1−3フコースとβ1−2キシロースが存在する(Johnson,K.D.およびChrispeels,M.J.,Plant Physiol,84,1301−1308,1897、Kimura,Y.ら,Biosci.Biotech.Biochem.,56,215−222,1992)。また、N結合型糖鎖としては植物糖鎖には動物糖鎖に見出されるシアル酸が存在せず、動物糖鎖に一般に存在するガラクトースも、シカモア細胞のラッカーゼの糖鎖に見出されているが(Takahashi,N.およびHotta,T.ら,Biochemistry,25,388−395,1986)、その例は少なく、またその結合型もβ1−3結合である(FEBS Lett 1997 9月 29,415(2)、186−191、植物細胞懸濁培養(Vaccinium mytillus L.)からの分泌ペルオキシダーゼ中のヒトLewis(a)炭水化物モチーフの同定、Melo NS、Nimtz M、Contradt HS、Fevereiro PS、Costa J;Plant J.1997 12月.12(6)1411−1417、Lewis aエピトープを保持するN−グリカンは、植物細胞の表面で発現される。Fitchette−Laine AC、Gomord V、Cabanes M、Michalski JC、Saint Macary M、Foucher B、Cavelier B、Hawes C、Lerouge P、Faye L)。この結合は、動物で見出される結合とは異なる。
【0008】
一般に、ヒトエリスロポエチン(EPO)などヒト由来の有用な糖タンパク質は、ヒトに近い糖鎖構造を有する糖タンパク質を生産する動物細胞宿主で生産されている。しかし、動物細胞で生産されたEPOは、本来の糖鎖構造とは異なる糖鎖構造を有し、生体内における活性が低下した(Takeuchi,M.ら,Proc.Natl.Acad.Sci.USA,86,7819−7822,1989)。ホルモン、インターフェロンなどのヒト由来の他の有用タンパク質の糖鎖構造が解析され、EPOと同じグルコシル化制限をともなって工業的に生産されている。
【0009】
近年、外来遺伝子を植物に導入する方法として、アグロバクテリウム法(Weising,K.ら,Annu.Rev.Genet.,22,421,1988)、エレクトロポレーション法(Toriyama,K.ら,Bio/Technology,6,1072,1988)、金粒子法(Gasser,C.G.およびFraley,R.T.、Science,244,1293,1989)などが確立され、アルブミン(Sijmons,P.C.ら,Bio/Technology,8,217,1990)、エンケファリン(Vandekerckhove,J.ら,Bio/Technology,7,929,1989)、およびモノクローナル抗体(Benvenulo,E.ら,Plant Mol.Biol.,17,865,1991およびHiatt,A.ら,Nature,342,76,1989)、が植物で生産され、そしてB型肝炎ウイルス主要表面抗原(HBsAg)(Mason,H.S.ら,Proc.Natl.Acad.Sci.USA.,89,11745,1992)、および分泌型IgA(Hiatt,A.およびMa,J.S.K.,FEBS Lett.,307,71,1992)を植物細胞中で生産し得ることも記載されている。しかし、ヒト由来の糖タンパク質を植物で発現する場合、植物は、ヒトと異なる糖鎖付加機構を有するため、生産される糖タンパク質の糖鎖は、ヒトで生産される糖鎖とは異なる構造を有し、本来の生理活性を示さず、また抗原となり得ることが指摘されている(Wilson I.B.H.ら,Glycobiol.,vol.8,No.7,pp651−661,1998)。
【発明の概要】
【課題を解決するための手段】
【0010】
本発明の目的は、哺乳動物、例えば、ヒト型の糖鎖をもつ植物が生産する組換え糖タンパク質を提供することにより、上記先行技術に関連する課題を解決することである。
【0011】
本発明は、ヒト型糖鎖をもつ糖タンパク質の生産方法に関する。この方法は、植物細胞に、非還元末端アセチルグルコサミン残基へのガラクトース残基の転移反応を行い得る酵素の遺伝子および異種糖タンパク質の遺伝子を導入して形質転換細胞を得る工程、および得られた形質転換植物細胞を培養する工程を包含する。
【0012】
本発明において、上記ヒト型糖鎖をもつ糖タンパク質は、コア糖鎖および外部糖鎖を含み、上記コア糖鎖は、複数のマンノースおよびアセチルグルコサミンから本質的になり、上記外部糖鎖は、非還元末端ガラクトースを含む末端糖鎖部分を含み得る。
【0013】
本発明において、上記外部糖鎖は、直鎖状構造または分岐状構造を有し得る。
【0014】
本発明において、上記分岐糖鎖部分は、モノ、バイ、トリ、またはテトラ構造であり得る。
【0015】
本発明において、上記糖タンパク質は、フコースおよびキシロースを含まなくともよい。
【0016】
本発明はまた、1つの局面で、非還元末端アセチルグルコサミン残基へのガラクトース残基の転移反応を行い得る糖鎖付加機構を備えた植物細胞に関し、この糖鎖付加機構は、コア糖鎖および外部糖鎖を含む糖鎖であって、このコア糖鎖は、複数のマンノースおよびアセチルグルコサミンから本質的になり、この外部糖鎖は非還元末端ガラクトースを含む末端糖鎖部分を含む糖鎖を付加する。
【0017】
本発明はまた、1つの局面で上記の方法によって得られたヒト型糖鎖をもつ糖タンパク質に関する。
【図面の簡単な説明】
【0018】
【図1】代表的なN結合型糖鎖の構造を示す模式図である。
【図2】hGTのクローニングの方法の模式図である。
【図3】hGT発現用ベクターpGAhGTの構築方法の模式図である。
【図4】形質転換体タバコ培養細胞のゲノムのサザン解析を示す写真である。図4(A)は、ゲノムDNA(40μg)をEcoRIおよびHindIIIで消化した後の電気泳動を示す。左側の数字はDNA分子量マーカーの位置を表す。図4(B)は、各形質転換細胞に組み込まれた、プロモーター、hGT、ターミネーターを含む2.2kbの断片の模式図を示す。
【図5】図5は、形質転換タバコBY2細胞(WT)および野生型タバコBY2細胞(WT)からの免疫反応性タンパク質のウェスタンブロッティングの写真である。タンパク質を変性させ、10% SDS−PAGEで電気泳動し、そしてニトロセルロース膜に電気的に転写した。レーンの数字は、サンプルがそれぞれ以下のものに由来したことを示す:1=GT1の細胞抽出物;2=GT6の細胞抽出物;3=GT8の細胞抽出物;4=GT9の細胞抽出物;5=野生型の細胞抽出物;6=GT1のミクロソーム画分;7=GT6のミクロソーム画分;8=GT8のミクロソーム画分;9=GT9のミクロソーム画分;および10=野生型のミクロソーム画分。
【図6】Ricinus communis(RCA120)アフィニティークロマトグラフィーを用いたガラクトシル化糖タンパク質の検出を示す電気泳動写真である。電気泳動ゲルは、銀染色により可視化した。レーン1および2は野生型BY2細胞からのタンパク質を示し、そしてレーン3および4は、形質転換GT6細胞からのタンパク質を示す。分子量はKDaの単位である。
【図7】Ricinus communis(RCA120)アフィニティークロマトグラフィーを用いたガラクトシル化糖タンパク質の検出を示すウェスタンブロッティングの写真である。電気泳動したゲルをニトロセルロース膜上へブロットした後、このブロットをレクチン(RCA120)染色して可視化した。レーン1および2は野生型BY2細胞からのタンパク質を示し、そしてレーン3および4は、形質転換GT6細胞からのタンパク質を示す。分子量はKDaの単位である。
【図8】Ricinus communis(RCA120)アフィニティークロマトグラフィーを用いたガラクトシル化糖タンパク質を、植物の複合型グリカンに存在するキシロースに特異的な抗血清でプローブしたブロッティングの写真である。レーン1および2は、BY2およびGT6からの総タンパク質抽出物をそれぞれ示し、レーン3は、RCA120アフィニティークロマトグラフィー後のGT6からの糖タンパク質を示す。分子量はKDaの単位である。
【図9】カナマイシン耐性遺伝子およびハイグロマイシン耐性遺伝子を有するバイナリーベクターであり、HRP cDNAを有するプラスミドpBIHm−HRPの模式図である。
【図10】トランスジェニック懸濁培養細胞におけるHRPの産生を示す等電点電気泳動およびウエスタンブロッティングの写真である。図10(A)は等電点電気泳動の結果を示し、そして図10(B)はウエスタンブロッティングの結果を示す。略語は以下の通りである:WT=野生型;BY2−HRP 1、5、および7=HRP遺伝子で形質転換したBY2細胞のクローン番号;ならびにGT6−HRP 4、5、および8=HRP遺伝子で形質転換したGT6細胞のクローン番号。
【図11】0.02%TFA中でアセトニトリルの濃度を0〜15%の直線状勾配で60分間かけて増加させ、1.2ml/分の流速で溶出したPA糖鎖の逆相HPLCパターンを示すグラフである。I〜XIは、サイズ分画HPLCにおいて溶出および精製した個々の画分を示す。励起波長および放出波長は、それぞれ、310nmおよび380nmであった。
【図12】図11におけるPA鎖のサイズ分画HPLCパターンを示すグラフである。PA糖鎖を、水−アセトニトリル混合物中で水の濃度を30〜50%に40分間かけて増加させ、0.8ml/分の流速で溶出させた。励起波長および放出波長は、それぞれ、310nmおよび380nmであった。
【図13】2つの標準品糖鎖AおよびBと比較した、逆相HPLCにおけるピーク−K2の溶出位置を示すグラフである。溶出条件は、図11と同様に0.02%TFA中でアセトニトリルの濃度を0〜15%の直線状勾配で60分間かけて増加させ、1.2ml/分の流速で溶出であった。
【図14】エクソグリコシダーゼ消化後に得られたガラクトシル化PA糖鎖のSF−HPLCプロフィールを示すグラフである。PA糖鎖を、水−アセトニトリル混合物中の水含量を30〜50%で25分間かけて増加させ、0.8ml/分の流速で溶出させた。(A)PA−糖鎖K−2;I.用いたガラクトシル化PA糖鎖の溶出位置;II.Iのβ−ガラクトシダーゼ消化物;III.IIのN−アセチル−β−D−グルコサミニダーゼ消化物;IV.IIIのタチナタマメ α−マンノシダーゼ消化物。(B)PAー糖鎖L;I.用いたガラクトシル化PA糖鎖の溶出位置;II.Iのβ−ガラクトシダーゼ消化物;III.IIのN−アセチル−β−D−グルコサミニダーゼ消化物;IV.IIIのα1−2マンノシダーゼ消化物;V.IIIのタチナタマメ α−マンノシダーゼ消化物。
【図15】形質転換細胞から得られたN結合型グリカンの推定構造を示す図である。括弧内の数字は、モル比を示す。
【図16】糖付加HRPの検出を示す、Ricinus communis 120 agglutinin(RCA120)アフィニティークロマトグラフィーの写真である。図16(A)は銀染色、そして図16(B)はレクチンRCA120での染色の結果である。レクチン染色のフィルターを細片に切断し、そして緩衝液単独(IおよびII)または過剰なガラクトースを含有する緩衝液(III)でプレインキュベートしたレクチンRCA120でプローブした。(II)では、HRPを、SDS−PAGEの前にDiplococcus pneumoniaeのβ−ガラクトシダーゼで処理した。レーン1はBY2−HRPからの収集画分;そしてレーン2はGT6−HRPからの収集画分である。左側の数字は、タンパク質の標準品の位置およびサイズ(KDa)を示す。
【図17】RCA120アフィニティークロマトグラフィーの後に精製されたHRPからのPA糖鎖の逆相HPLCの結果を示す図である。
【図18】植物特異的な複合型グリカンの免疫検出を示すウェスタンブロッティングの写真である。精製されたHRPをSDS−PAGEで分画し、ニトロセルロースに転写し、そしてウサギ抗HRP(A)または植物の複合型グリカンに特異的な抗血清(B)でプローブした。レーン1=BY2−HRPからの精製HRP;レーン2=RCA120アフィニティークロマトグラフィー後のGT6−HRPからのガラクトシル化HRP。分子のサイズマーカーの位置を、KDaで左に示す。形質転換体GT6−HRP細胞由来のHRP上のガラクトシル化N−グリカンは、植物N−グリカンの指標であるβ1,2キシロース残基と特異的に反応することが示された抗血清と反応しなかった。
【図19】小胞体およびゴルジ体中の糖鎖修飾経路に関与するN−結合グリカンの構造および酵素。白抜き菱形はグルコースを、白抜き四角はGlcNAcを、白抜き丸はマンノースを、黒丸はガラクトースを、そして黒四角はシアル酸をそれぞれ示す。
【図20】GT6細胞株中のN−結合グリカン類の構造、および各N−結合グリカンの比率を、同様に測定された野生型BY2細胞株それらとともに示す。白抜き四角はGlcNAcを、白抜き丸はマンノースを、黒丸はガラクトースを、そして黒四角はシアル酸をそれぞれ示す。
【図21】本発明の1つの実施態様を図示する。GT6細胞株において、アイソマー類Man7−、Man6−およびMan5GlcNAc2が認められた。これらの高マンノース型オリゴサッカライドは、いくつかのグリカンプロセッシング酵素によって、β1,4−ガラクトシルトランスフェラーゼ(Gal T)の基質であるように変換されるので、GlcNAc I、Man IおよびMan II cDNA類の導入は、オリゴサッカライドMan7−5GlcNAc2を、GalTの基質であり得るGlcNAcMan3GlcNAc2により効率的に導き得る。
【図22】この図もまた、本発明の1つの実施態様を示す。1,4−ガラクトシルトランスフェラーゼ(Gal T)は、UDP−ガラクトースをドナー基質として用い、そしてGlcNAc2Man3GlcNAc2をアクセプター基質として用いる。UDP−ガラクトースのオリゴサッカライドを産生し得る。効率的な供給は、Gal T酵素反応を増大し、そしてより多くのガラクトシル化オリゴサッカライドを産生し得る。
【発明を実施するための形態】
【0019】
以下、本発明を詳細に説明する。
【0020】
本発明を実施することにおいて、他に特定されない限り、当該分野で公知である、タンパク質の分離および分析法、ならびに免疫学的手法が採用され得る。これらの手法は、市販のキット、抗体、標識物質などを使用して行い得る。
【0021】
本発明の方法は、ヒト型糖鎖をもつ糖タンパク質の生産方法に関する。本明細書において、「ヒト型糖鎖」とは、N−アセチルグルコサミン残基と結合したガラクトース残基を有する糖鎖をいう。ヒト型糖鎖におけるガラクトース残基は糖鎖の末端であってもよいし、ガラクトース残基のさらに外側にシアル酸残基が結合していてもよい。本発明の糖タンパク質は、ヒト型糖鎖のコア糖鎖部分、分岐糖鎖部分、および末端糖鎖部分からなる1つ以上の部分において、キシロースおよびフコースの少なくとも一方が結合していないことが好ましく、ヒト型糖鎖のいずれの部分においてもキシロースおよびフコースの少なくとも一方が結合していないことがより好ましく、最も好ましくはヒト型糖鎖中にキシロースおよびフコースのいずれも含まないことがさらにより好ましい。
【0022】
植物細胞とは、所望の任意の植物細胞であり得る。植物細胞は、培養細胞、培養組織中の細胞、培養器官中の細胞、または植物体中の細胞のいずれの形態であってもよい。好ましくは、植物細胞は、培養細胞、培養組織中の細胞、または培養器官中の細胞であり、最も好ましくは、この植物細胞は、ヒト型糖鎖をもつ糖タンパク質を産生する、完全な植物、またはその一部にある細胞である。本発明の生産方法に使用され得る植物種は、遺伝子導入を行い得る任意の植物種であり得る。本発明の生産方法に使用され得る植物種の例としては、ナス科、イネ科、アブラナ科、バラ科、マメ科、ウリ科、シソ科、ユリ科、アカザ科、セリ科の植物が挙げられる。
【0023】
ナス科の植物の例としては、Nicotiana、Solanum、Datura、Lycopersion、またはPetuniaに属する植物が挙げられ、例えば、タバコ、ナス、ジャガイモ、トマト、トウガラシ、ペチュニアなどを含む。
【0024】
イネ科の植物の例としては、Oryza、Hordenum、Secale、Scccharum、Echinochloa、またはZeaに属する植物が挙げられ、例えば、イネ、オオムギ、ライムギ、ヒエ、モロコシ、トウモロコシなどを含む。
【0025】
アブラナ科の植物の例としては、Raphanus、Brassica、Arabidopsis、Wasabia、またはCapsellaに属する植物が挙げられ、例えば、大根、アブラナ、シロイヌナズナ、ワサビ、ナズナなどを含む。
【0026】
バラ科の植物の例としては、Orunus、Malus、Pynus、Fragaria、またはRosaに属する植物が挙げられ、例えば、ウメ、モモ、リンゴ、ナシ、オランダイチゴ、バラなどを含む。
【0027】
マメ科の植物の例としては、Glycine、Vigna、Phaseolus、Pisum、Vicia、Arachis、Trifolium、Alphalfa、またはMedicagoに属する植物が挙げられ、例えば、ダイズ、アズキ、インゲンマメ、エンドウ、ソラマメ、ラッカセイ、クローバ、ウマゴヤシなどを含む。
【0028】
ウリ科の植物の例としては、Luffa、Cucurbita、またはCucumisに属する植物が挙げられ、例えば、ヘチマ、カボチャ、キュウリ、メロンなどを含む。
【0029】
シソ科の植物の例としては、Lavandula、Mentha、またはPerillaに属する植物が挙げられ、例えば、ラベンダー、ハッカ、シソなどを含む。
【0030】
ユリ科に属する植物の例としては、Allium、Lilium、またはTulipaに属する植物が挙げられ、例えば、ネギ、ニンニク、ユリ、チューリップなどを含む。
【0031】
アカザ科の植物の例としては、Spinaciaに属する植物が挙げられ、例えば、ホウレンソウを含む。
【0032】
セリ科の植物の例としては、Angelica、Daucus、Cryptotaenia、またはApitumに属する植物が挙げられ、例えば、シシウド、ニンジン、ミツバ、セロリなどを含む。
【0033】
本発明の生産方法に用いられる植物は、好ましくはタバコ、トマト、ジャガイモ、イネ、トウモロコシ、ダイコン、ダイズ、エンドウ、ウマゴヤシ、およびホウレンソウであり、より好ましくは、タバコ、トマト、ジャガイモ、トウモロコシ、およびダイズである。
【0034】
本明細書において、「非還元末端アセチルグルコサミン残基へのガラクトース残基の転移反応を行い得る酵素」とは、植物細胞内の糖タンパク質のタンパク質部分の合成後、糖鎖付加の際に生じる非還元末端アセチルグルコサミン残基へガラクトース残基を転移し得る酵素である。このような酵素の例としては、ガラクトシルトランスフェラーゼ、ラクトースシンターゼ、β−ガラクトシダーゼが挙げられる。このような酵素は、任意の動物種に由来し得るが、哺乳動物に由来することが好ましく、ヒトに由来することがより好ましい。
【0035】
本明細書において、「非還元末端アセチルグルコサミン残基へのガラクトース残基の転移反応を行い得る酵素の遺伝子」は、この酵素をコードすることが公知のヌクレオチド配列を用いて任意の動物細胞から単離してもよいし、市販のものを購入してもよいし、これらを植物での発現に適切なように改変して用いてもよい。
【0036】
本明細書では、「遺伝子」とは、構造遺伝子部分をいう。遺伝子には、植物での発現に適切なように、プロモーター、オペレーター、およびターミネーターなどの制御配列が連結され得る。
【0037】
本明細書において、「異種糖タンパク質」とは、本発明に用いられる植物において本来発現されない糖タンパク質をいう。異種糖タンパク質の例としては、酵素、ホルモン、サイトカイン、抗体、ワクチン、レセプター、血清タンパク質などが挙げられる。酵素の例としては、西洋ワサビペルオキシダーゼ、キナーゼ、グルコセレブロシダーゼ(glucocerebrosidase)、α−ガラクトシダーゼ、フィターゼ、TPA(tissue−type plasminogen activator)、HMG−CoAレダクターゼ(HMG−CoA reductase)などが挙げられる。ホルモンおよびサイトカインの例としては、エンケファリン、インターフェロンアルファ、GM−CSF、G−CSF、絨毛性性腺刺激ホルモン、インターロイキン−2、インターフェロン−ベータ、インターフェロン−ガンマ、エリスロポイエチン、血管内皮細胞増殖因子(vascular endothelial growth factor)、ヒト絨毛性ゴナドトロピン(HCG)、黄体形成ホルモン(LH)、甲状腺刺激ホルモン(TSH)、プロラクチン、卵胞刺激ホルモンなどが挙げられる。抗体の例としては、IgG、scFvなどが挙げられる。ワクチンの例としては、B型肝炎表面抗原、ロタウイルス抗原、大腸菌エンテロトキシン、マラリア抗原、狂犬病ウイルスrabies virusのGタンパク質、HIVウイルス糖タンパク質(例えば、gp120)などが挙げられる。レセプターおよびマトリックスタンパク質の例としては、EGFレセプター、フィブロネクチン、α1−アンチトリプシン、凝固因子VIIIなどが挙げられる。血清タンパク質の例としては、アルブミン、補体系タンパク質、プラスミノーゲン、コルチコステロイド結合グロブリン(corticosteroid−binding globulin)、スロキシン結合グロブリン(Throxine−binding globulin)、プロテインC(protein C)などが挙げられる。
【0038】
本明細書において、「異種糖タンパク質の遺伝子」は、目的の異種糖タンパク質をコードすることが公知のヌクレオチド配列を用いて任意の細胞から単離してもよいし、市販のものを購入してもよいし、これらを植物での発現に適切なように改変して用いてもよい。
【0039】
非還元末端アセチルグルコサミン残基へのガラクトース残基の転移反応を行い得る酵素および異種糖タンパク質の遺伝子は、当該分野で公知の方法により、植物細胞へ導入される。これらの遺伝子は、別々に導入してもよいし、同時に導入してもよい。植物細胞への遺伝子の導入方法の例としては、アグロバクテリウム法、エレクトロポレーション法、金粒子法などが挙げられる。
【0040】
遺伝子が導入された植物細胞は、当該分野で公知の方法により、導入された遺伝子の発現が確認され得る。このような確認方法としては、銀染色、ウェスタンブロッティング、ノザンハイブリダイゼーション、酵素活性の検出などが挙げられる。導入された遺伝子を発現する細胞は、形質転換細胞である。
【0041】
非還元末端アセチルグルコサミン残基へのガラクトース残基の転移反応を行い得る酵素および異種糖タンパク質を発現する形質転換細胞は、ヒト型の糖鎖を有する異種糖タンパク質を発現する。つまり、このようにして得られた形質転換植物は、ヒト型の糖鎖付加機構を有する。形質転換細胞を培養することにより、ヒト型の糖タンパク質が大量に生産され得る。ヒト型の糖タンパク質は、コア糖鎖および外部糖鎖を含み、このコア糖鎖は、複数のマンノースおよびアセチルグルコサミンから本質的になる。得られる糖タンパク質の外部糖鎖は、非還元末端糖鎖部分を含む。外部糖鎖は、直鎖状構造をもっていても分岐状構造をもっていてもよい。分岐糖鎖部分が、モノ、バイ、トリ、またはテトラ構造のいずれかであり得る。形質転換細胞により生産される糖タンパク質は、好ましくは、フコースまたはキシロースを含まない。
【0042】
得られた形質転換植物細胞は、培養細胞の状態で維持されてもよいし、特定の組織または器官へと分化させてもよいし、完全な植物体に再生させてもよい。あるいは、完全な植物体から得られる、種子、果実、葉、根、茎、花などの部分であってもよい。
【0043】
形質転換植物細胞により生産された、ヒト型の糖鎖をもつ糖タンパク質は、植物細胞から単離または抽出されてもよい。糖タンパク質の単離方法は、当該分野で公知である。あるいは、本発明の糖タンパク質は、形質転換細胞中に含まれたままの状態で食用に供され得る。本発明の糖タンパク質は、ヒト型の糖鎖付加を有するので、抗原性を有さず、それゆえ、ヒトを含む動物への投与に適している。
【0044】
以後、本発明を、制限ではない例示の実施例によって詳細に記載する。
【実施例】
【0045】
(実施例1)ヒトβ1−4ガラクトース転移酵素遺伝子のクローニング
β1−4ガラクトース転移酵素(hGT)(EC2.4.1.38)は、すでにクローン化されており、400アミノ酸からなる一次構造が明らかにされている(Masri,K.A.ら,Biochem.Biophys.Res.Commun.,157,657−663,1988)。
【0046】
(1)プライマーの作製と鋳型DNA
Masri.らの報告を参考にし、以下のようなプライマーを作製した。
hGT−5Eco:5’−AAAGAATTCGCGATGCCAGGCGCGCGTCCCT−3’(配列番号1)
hGT−2Sal:3’−TCGATCGCAAAACCATGTGCAGCTGATG−5’(配列番号2)
hGT−7Spe:3’−ACGGGACTCCTCAGGGGCGATGATCATAA−5’(配列番号3)
hGT6Spe:5’−AAGACTAGTGGGCCCCATGCTGATTGA−3’(配列番号4)
鋳型DNAは、Clontech社から購入したヒトゲノムDNA、ヒト胎盤cDNA、ヒト腎臓cDNAを用いた。
【0047】
(2)hGT遺伝子cDNAのクローニング
(i)ヒトゲノムDNAを鋳型とし、hGT−5EcoとhGT−7Speとをプライマーに用い;および(ii)ヒト胎盤cDNAを鋳型とし、hGT−2SalとhGT6Speとをプライマーに用いた、2つの組み合わせで以下の条件でPCR反応を行い、hGTをコードする領域を含む0.4kbおよび0.8kbの断片を得た。
(PCR反応系)鋳型DNA1μl、10×PCR緩衝液5μl、dNTPs(200μM)4μl、プライマー(10pmol)、Tagポリメラーゼ(宝酒造(株))0.5μl(Tubポリメラーゼの場合は0.2μl)に水を加えて50μlとした。
(PCR反応条件)第1段階:サイクル数1、変性(94℃)5分、アニーリング(55℃)1分、伸長(72℃)2分。第2段階:サイクル数30、変性(94℃)1分、アニーリング(55℃)1分、伸長(72℃)2分。第3段階:サイクル数1、変性(94℃)1分、アニーリング(55℃)2分、伸長(72℃)5分。
【0048】
得られた2つの断片を組み合わせてhGT遺伝子cDNAを構築し、pBluescriptIISK+(SK)にサブクローニングした。pBluescriptIISK+(SK)は、Stratagene社から購入した。図2に、hGT遺伝子cDNAを含むプラスミドの構築を示す。得られたhGT遺伝子の塩基配列を配列番号5に、そして推定されるアミノ酸配列を配列番号6に示す。
【0049】
得られた配列は、Masriら(上述)に開示のhGT配列と比較して以下の点で異なっていた。a)位置528のAがGに、位置562のCがTに、位置1047のAがGにそれぞれ変わっていたがコードされるアミノ酸に変化はなかった。b)位置622から630の9塩基が欠失していた。c)得られた上記の0.4kbおよび0.8kbの断片を接続するためにプライマー作製時に位置405のGをAに、位置408のTをAにそれぞれ変換した。なお、hGT遺伝子cDNAには2つの開始コドン(ATG)があるが、本実験では、第2番目の開始コドン(位置37)から翻訳が始まるように設計した。
【0050】
(実施例2)hGT遺伝子のタバコ培養細胞への導入
(1)hGTは大腸菌で活性型として発現することが報告されている(Aoki, D.ら,EMBO J.,9,3171,1990、およびNakazawa,K.ら,J.Biochem.,113,747,1993)。
【0051】
hGTをタバコ培養細胞で発現するために、発現用ベクターpGAhGTを図3に示すように構築した。プロモーターとしては、植物細胞内で構成的に発現するカリフラワーモザイクウイルス35Sプロモーター(CaMV 35Sプロモーター)を用いた。選択マーカーにはカナマイシン耐性遺伝子を用いた。pGAhGTをアグロバクテリウムを介してタバコ培養細胞に導入した。
【0052】
アグロバクテリウムの形質転換は、Bevan.ら,のtriparental mating法(Bevan,M.,Nucleic Acid Res.,12,8711,1984)を用いて行った。pGA系プラスミド(An,G.,Methods Enzymol.153,292,1987)をもつ大腸菌Escherichia coli DH5α株(suE44、ΔlacU169、(φ80lacZΔM15)、hsdR17)(Bethesda Research Laboratories Inc.:Focus 8(2)、9(1986))、およびヘルパープラスミドpRK2013(Bevan,M.,Nucleic Acid Res.,12,8711,1984)をもつ大腸菌Escherichia coli HB101を、それぞれ12.5mg/lのテトラサイクリン、50mg/lのカナマイシンを含む2×YT培地で37℃で1晩、アグロバクテリウムAgrobacterium tumefaciens EHA101株(Elizanbeth,E.H.,J.Bacteriol.,168,1291,1986)を、50mg/lのカナマイシン、25mg/lのクロラムフェニコールを含む2×YT培地で28℃で2晩培養した。各培養液1.5mlをエッペンドルフチューブにとり集菌した後、LB培地で3回洗浄した。得られた菌体をそれぞれ100μlの2×YT培地に懸濁した後、3種類の菌を混合し、2×YT寒天培地に塗抹し、28℃で培養してpGA系プラスミドを大腸菌からアグロバクテリウムに接合伝達させた。2日後、2×YT寒天培地上で一面に増殖した菌体の一部を白金耳でかきとり、50mg/lのカナマイシン、12.5mg/lのテトラサイクリン、25mg/lのクロラムフェニコールを含むLB寒天培地上に塗布した。28℃で2日間培養した後、単一コロニーを選択した。
【0053】
タバコ培養細胞の形質転換は、An,G.,Plant Mol.Bio.Mannual,A3,1.に記載の方法により行った。テトラサイクリン12.5mg/lを含むLB培地で28℃で36時間培養したアグロバクテリウム(pGA系のプラスミドをもつEHA101株)と培養4日目のタバコ培養細胞Nicotiana tabacum L.cv.bright yellow 2(理化学研究所ライフサイエンス筑波研究センター、ジーンバンク室植物細胞開発銀行のカタログ番号RPC1より細胞株名BY−2として入手した)の懸濁液をそれぞれ100μl、4mlずつシャーレに入れてよく混ぜ、25℃に暗所で静置した。2日後シャーレの中の培養液を遠心管に移して遠心分離(1000rpm、5分)により上澄みを除いた。次に新しい培地を入れて遠心分離し、細胞を150〜200mg/lのカナマイシン、250gmg/lのカルべニシリンの入った改変LS寒天培地のプレートに塗抹し、25℃暗黒下で静置した。約2〜3週間後にカルス化した細胞を新しいプレートに移植し、増殖しているクローンを選択した。さらに2〜3週間後に、カナマイシン、カルベニシリンを加えた改変LS培地30mlに移し、継代培養を行った。約1ケ月間選択を繰り返した。得られたいくつかの耐性株の中から無作為に6つの耐性株(GT1、4、5、6、8、および9)を選択した。
【0054】
(2)導入されたhGT遺伝子の確認
得られた耐性株についてT−DNA中の、CaMV35Sプロモーター−hGT遺伝子cDNA−NOSターミネーターを含む2.2kbの断片がタバコ培養細胞のゲノムDNA中に組み込まれていることをサザン解析により確認した。上記の各耐性株からゲノムDNAを調製し、EcoRI、HindIIIで消化した後、サザン解析を行った。
【0055】
タバコ培養細胞からの染色体DNAの調製は、渡部の方法(渡部格、「クローニングとシーケンス」、植物バイオテクノロジー実験マニュアル、農村分化社)に従って行った。まず最初に、タバコ培養細胞10mlを液体窒素で凍結し、乳鉢、乳棒を用いて、粉末状になるまで粉砕した。得られた約5gの粉末が融解しないうちに、遠心管(40ml)中で60℃に予熱した5mlの2×CTAB(セチルトリメチルアンモニウムブロミド(cetyltrimethylammonium bromide))液に入れてゆっくりよく混ぜ、60℃で10分間以上、時々混ぜながら保温した。クロロホルム:イソアミルアルコール(24:1)5mlを加え、エマルジョンができるまでよく混ぜてから、遠心分離(2,800rpm、15分、室温)した。上層を新しい40ml遠心管に移し、クロロホルム:イソアミルアルコール(24:1)を用いて抽出操作を繰り返した。得られた上層に1/10容量の10%CTABを入れてよく混合した後、遠心分離操作を行った(2,800rpm、15分、室温)。上層を新しい遠心管に移し、1容量の冷イソプロパノールを入れて良く混ぜ、遠心分離(4,500rpm、20分、室温)した。上清液をアスピレータで除いた後、5mlの1M塩化ナトリウムを含むTE緩衝液を入れ、55〜60℃で完全に溶解した。これに、5mlの冷イソプロパノールを入れ、DNAが見えたら、チップの先に引っかけてエッペンドルフチューブ(80%冷エタノール入り)に移しリンスした。さらにDNAを70%エタノールでリンスし、乾燥した沈殿を、適当量のTE緩衝液に溶かし、5μlのRNAaseA(10mg/ml)を加え、37℃で1時間反応させた:2×CTAB液の組成;2%CTAB、0.1M Tris−HCl(pH8.0)、1.4M塩化ナトリウム、1%ポリビニルピロリドン(PVP);10%CTAB液の組成;10%CTAB、0.7M塩化ナトリウム。
【0056】
サザン解析は、以下のように行った。
【0057】
(i)DNAの電気泳動およびアルカリ変性:得られた染色体DNA40μgを制限酵素で完全分解した後、標準的な方法を用い、1.5%アガロースゲル電気泳動(50V)を行った。ゲルをエチジウムブロマイドで染色し、写真撮影した後、400mlの0.25M HCl中で20分間振とうした後、液を捨て、400mlの変性溶液(1.5M NaCl、0.5M NaOH)にゲルを浸し、45分間ゆっくり振とうした。次いで液を捨て、400mlの中和溶液(1.5M NaCl、0.5M Tris−Cl(pH7.4))を入れて15分間ゆっくり振とうした後液を捨て、再び中和溶液を400ml入れて15分間ゆっくり振とうした。(ii)トランスファー:電気泳動後のDNAを、20×SSCを用いてナイロンメンブレン(Hybond−N Amersham)にトランスファーした。トランスファーは12時間以上行った。プロットしたメンブレンを、室温で、1時間乾燥させた後、5分間UV固定を行った。20×SSCの組成:3M NaCl、0.3Mクエン酸ナトリウム。(iii)DNAプローブの調製:DNAプローブの調製は、Random prime Labeling Kit(宝酒造)を用いて行った。エッペンドルフチューブに次の反応液を調製し、95℃、3分間加熱後、氷中で急冷した;鋳型DNA 25ng、Random Primer 2μl、水を加えて5μlとする。10×緩衝液、dNTPをそれぞれ2.5μl、[α−32P]dCTP(1.85MBq、50mCi)を5μl加え、H2Oで24μlにフィルアップした。Klenow fragment 1μlを加え、37℃で10分間保温した後、NAP10カラム(Pharmacia社製)で溶出させてDNAを精製した。95℃で3分間加熱した後、氷中で急冷し、ハイブリダイゼーションブローブとした。(iv)ハイブリダイゼーション:0.05mg/ml 0.5%(w/v)SDSを、以下のプレハイブリダイゼーション溶液に加えて、上記(ii)のメンブレンを浸漬し、42℃で2時間以上プレハイブリダイゼーションを行った。その後、(iii)で調製したDNAプローブを加え、42℃で12時間以上ハイブリダイゼーションを行った:プレハイブリダイゼーション溶液の組成;5×SSC、50mMリン酸ナトリウム、50%(w/v)ホルムアミド、5×デンハルト溶液(100×デンハルト溶液を希釈して使用)、0.1%(w/v)SDS。100×デンハルト溶液の組成:2%(w/v)BSA、2%(w/v)Ficol 400、2%(w/v)ポリビニルピロリドン(PVP)。(v)オートラジオグラフィー:以下の順で洗浄した後、標準的な方法によりオートラジオグラフィーを行った。2×SSC、0.1%SDS中、65℃で15分間、2回、次いで0.1×SSC、0.1%SDS中、65℃で15分間、1回。
【0058】
上記で得られた耐性株のそれぞれから調製したゲノムDNAのサザン分析の結果を図4に示す。図4に示されるように、GT1、6、8および9の4株についてhGT遺伝子が組み込まれていることが確認された。
【0059】
(実施例3)ガラクトシルトランスフェラーゼ形質転換体の解析
培養5〜7日目の形質転換体(GT−1、6、8、および9)および野生型のBY−2細胞の培養液からそれぞれ細胞を回収し、抽出緩衝液(25mM Tris−HCl、pH7.4;0.25Mスクロース、1mM MgCl2、50mM KCl)に懸濁し、そして超音波処理により破壊(200W;Kaijo Denki Co.,Japan)またはホモゲナイズした。細胞抽出液とミクロソーム画分を、Schwientek,T.らの方法(Schwientek,T.およびErnst,J.F.,Gene145,299−303,1994)に従って調製した。hGTタンパク質の発現は、抗ヒトガラクトシルトランスフェラーゼ(GT)モノクローナル抗体(MAb 8628;1:5000)(Uejima,T.ら,Cancer Res,52,6158−6163,1992;Uemura,M.ら,Cancer Res.,52,6153−6157,1992)(創価大学成松博士より供与)を用いてウェスタンブロッティングにより検出した。次いでブロットを西洋ワサビペルオキシダーゼ結合ヤギ抗マウスIgG(5%スキムミルク中1:1000;EY Laboratories,Inc.,CA)とインキュベートし、洗浄し、そして西洋ワサビペルオキシダーゼ発色反応は、POD免疫染色キット(Wako Chemicals,Osaka)を用いて実施した。
【0060】
植物特異的複合グリカンのイムノブロット分析は、ニンジン細胞壁β−フルクトシダーゼに対して惹起されたポリクローナル抗血清および西洋ワサビペルオキシダーゼ結合ヤギ抗ウサギIgG抗体(5%スキムミルク中1:1000;Sigma)を用いて実施した(Lauriere,M.ら,Plant Physiol.90,1182−1188,1989)。
【0061】
β1−4ガラクトシルトランスフェラーゼ活性は、基質として、UDP−ガラクトースおよびピリジルアミノ標識(PA−)GlcNAc2Man3GlcNAc2(GlcNAc2Man3GlcNAc2−PA)を用いてアッセイした(Morita,N.ら,J.Biochem.103,332−335,1988)。酵素反応液は、1〜120μgのタンパク質、25mMカコジル酸ナトリウム(pH7.4)、10mM MnCl2、200μM UDP−ガラクトース、および100nM GlcNAc2Man3GlcNAc2−PAを含んでいた。反応産物は、PALPAK Type RおよびType Nカラム(Takara社製)を製造業者の推奨する処方に従って用いてHPLCにより分析した。PA標識した標準として用いたGlcNAc2Man3GlcNAc2−PA、Gal2GlcNAc2Man3GlcNAc2−PAおよびGalGlcNAc2Man3GlcNAc2−PAの2つのアイソマーは、TAKARA SHUZOおよびHonen,Co.から購入した。
【0062】
図5に、形質転換体および野生型由来のタンパク質のイムノブロッティングを示す。図5に示すように、分子量約50kDaのポジティブシグナルが観察された。これは、アミノ酸配列から推定される分子量(約40kDa)より大きく、腹水から精製された、または酵母で発現されたウシガラクトシルトランスフェラーゼ(Uemura,M.ら,Cancer Res.52,6153−6157,1992;Schwientek,T.ら,J.Biol.Chem.271(7),3398−3405,1996)とほぼ等しかった。細胞溶解液(図5、レーン1〜4)と比較してミクロソーム画分(図5、レーン6〜8)で観察された強い免疫反応性バンドは、hGTが細胞内に優先的に局在化していたことを示す。野生型細胞には、免疫反応性のバンドは検出されなかった。
【0063】
形質転換体GT6および野生型BY−2のミクロソーム画分のタンパク質を、RCA120アガロースカラム(Wako Chemicals,Osaka)に結合させ、15容量の10mM酢酸アンモニウムpH6.0で洗浄した。次いで0.2Mラクトースで結合したタンパク質を溶出し、SDS−PAGEで分画した後、銀染色(Wako Silver Staining Kit)(図6)またはレクチン染色した(図7)。レクチン染色には、膜ブロットをTTBS緩衝液(10mM Tris−HCl、pH7.4;0.15M NaCl、0.05%Tween20)中で洗浄し、西洋ワサビペルオキシダーゼ標識RCA120(Honen Co.社製)とインキュベートし、そしてPOD免疫染色キット(Wako Chemicals,Osaka)を用いてガラクトシル化されたグリカンを可視化した(図7)。図7に示すように、野生型のBY2細胞ではRCA120の結合は観察されなかったが、GT6は、グリカン成分の非還元末端にガラクトースを持つ糖タンパク質を有していた。
【0064】
野生型BY2細胞およびGT6細胞からのタンパク質抽出物およびRCA120アフィニティークロマトグラフィーからのGT6の溶出タンパク質を、複合グリカンに特異的なポリクローナル抗血清でプローブした(図8)。用いた抗血清は主に植物糖タンパク質上のβ1,2−キシロース残基に結合する(Lauriere,M.ら,Plant Physiol.90,1182−1188,1989)。図8に示すように、野生型BY2細胞(レーン1)は用いたポリクローナル抗血清と反応するいくつかの糖タンパク質を含んでいた。GT6は抗血清と反応する糖タンパク質の量が減少した(レーン2)。RCA120アフィニティークロマトグラフィーから溶出したGT6糖タンパク質は、ポリクローナル抗血清に結合せず、ガラクトシル化されたグリカンはβ1,2−キシロース残基を含んでいないことを示した(レーン3)。
【0065】
(実施例4)hGT導入タバコ培養細胞への西洋ワサビペルオキシダーゼ(HRP)遺伝子の導入
上記で得られたGT6株に西洋ワサビペルオキシダーゼを導入した。異なる型の植物ペルオキシダーゼの中で、西洋ワサビペルオキシダーゼ、特に中性アイソザイムCは、いわゆるHRP(EC1.11.1.7.)として、最もよく研究されている。HRPは、基質特異性が広く、安定性に優れることから種々の酵素反応、例えば、ウェスタンブロッティングの2次抗体などの酵素免疫法に利用されている。現在、西洋ワサビペルオキシダーゼアイソザイム遺伝子は多数クローニングされている(Fujiyama,K.ら,Eur.J.Biochem.,173,681−687,1988およびFujiyama,K.ら,Gene,89,163−169,1990)。それらのうちprxClaがコードするClaペルオキシダーゼ(ClaPRX)は、N末端側の30アミノ酸、C末端側の15アミノ酸からなるエキストラペプチドを含む353アミノ酸からなるタンパク質として翻訳され、その後プロセッシングをうけて、308アミノ酸からなる成熟型酵素となることが明らかにされた(Fujiyama,K.ら,Eur.J.Biochem.,173,681−687,1988)。ClaPRXの分子量は、42,200〜44,000であり、そのうち糖鎖が22〜27%を占めており、N−結合型糖鎖は8本存在することが知られている(Welinder,K.G.,Eur.J.Biochem.,96,483−502,1979)。ClaPRX遺伝子の導入は、図9に示すHRP発現用バイナリーベクターpBIHm−HRPを用いて行った。
【0066】
なお、このpBIHm−HRPは、以下のようにして調製した。まず、HRPcDNAを保持する植物用発現ベクター35S−prxC1a(KawaokaA.ら,J.Ferment.Bioeng.,78,49−53,1994)から1.9kbpのHindIII−SacI断片を切り出して調製した。このHindIII−SacI断片は、0.8kbpのCaMV35Sプロモーターに続く完全長の1.1kbpのprxC1acDNA断片を含む。1.9kbpのHindIII−SacI断片を、バイナリーベクターであるpBI101HmB(Akama K.ら,Plant Cell Rep.12,7−11,1992)のHindIII−SacI部位に挿入した。HPT遺伝子(ハイグロマイシン耐性遺伝子)の3’末端側のBamHI部位は破壊してある。
【0067】
GT6株はカナマイシン耐性であるので、選択マーカーとしてハイグロマイシン耐性遺伝子であるhpt遺伝子(Griz,L.およびDavies,J.、Gene,25,179−188,1983)を用いた。HRPによるGT6株の形質転換は、Rempel.D.H.およびNelson,L.M.の方法(Rempel,H.C.およびNelson,L.M.、Transgenic Res.4:199−207,1995)に記載の方法により行った。同様に、コントロールのHRP形質転換体を得るために、野生型BY2細胞にHRP形質転換体を導入して、BY2−HRP株を得た。hGTとHRPの二重形質転換体GT6−HRPは、通常の形質転換体と同様の期間で取得することができた。
【0068】
(実施例5)二重形質転換タバコ培養細胞におけるHRPの発現の確認
得られた二重形質転換体GT6−HRP、コントロールのBY2−HRP、野生型(WT)について、以下に示す方法で、HRP活性発現について調べた。表1に示すように、HRP遺伝子導入形質転換体は、野生株に比べて、約5倍のペルオキシダーゼ活性を示した。
【0069】
【表1】
【0070】
野生型にHRP遺伝子を導入して得られたクローンBY2−HRPは、hGTとHRPの二重形質転換体GT6−HRPと同程度のペルオキシダーゼ活性を示した。
【0071】
(ペルオキシダーゼ活性測定)
タバコ培養細胞をSolution Dの入ったエッペンドルフチューブに入れ、ホモジナイザー(HomogenizerS−203、池田理化社製)を用いて粉砕した。遠心分離(12,000rpm、20分間、4℃)により上清液を回収し、これを粗酵素液とした。1mlのSolution A、1mlのSolution Bおよび2mlのSolution Cを混合し、25℃で5分間保温した。これにSolution Dで適当に希釈した粗酵素液を加え、25℃で3分間反応させた。0.5mlの1N HClを加えることにより反応を停止し、480nmの吸光度を測定した。対照としては、酵素添加前に1N HClを添加した液を用いた:
Solution A;1mM ο−aminophenol
Solution B;4mM H2O2
Solution C;200mM リン酸ナトリウム緩衝液(pH7.0)
Solution D;10mM リン酸ナトリウム緩衝液(pH6.0)
【0072】
次に、このペルオキシダーゼ活性の上昇が、HRPの発現によるものであるか否かを確認するために、等電点電気泳動で分離後、ゲルの活性染色を行った。等電点電気泳動は、BIO−RAD社製のモデル111ミニIEFセルを用いて行った。PAGEゲルサポートフィルムの疎水面をガラスプレートに密着させた後、キャスティングトレーにのせた。調製したゲル用溶液をサポートフィルムとキャスティングトレーの間に流し込んで、蛍光灯の下45分間光重合させた。作製したゲルに試料を塗布し、蒸留水で湿らせた泳動槽にある両グラファイト電極にゲルが接するように置き、100V 15分、200V 15分、450V 60分の条件で電気泳動を行った。ゲル用溶液(ゲル1枚分)の組成は以下の通りである:蒸留水2.75ml、アクリルアミド(25%T、3%C)1.0ml、25%グリセロール1.0ml、バイオライト(40%、pH3〜10)0.25ml、10%過硫酸アンモニウム7.5μl、0.1%リボフラビン−5’−リン酸ナトリウム25μl、TEMED 1.5μl。
【0073】
ペルオキシダーゼの活性染色は、関根らの方法(関根ら,植物細胞工学、6、71−75、1994)に従って行った。図10に示すように、BY2−HRP株と、GT6−HRP株において、野生株には存在しない顕著なバンドがpI7.8の位置に検出された。抗HRP抗体を用いたウェスタン解析を行った結果、同じpI7.8に対応する位置にシグナルが検出され、hGTとHRPの二重形質転換体GT6−HRPにおいてHRPが発現していることが確認された。
【0074】
(実施例6)形質転換細胞GT6細胞中のN−結合型糖鎖の構造解析
(糖鎖構造の解析方法)
形質転換細胞GT6細胞のN−結合型糖鎖構造を、逆相HPLCとサイズ分画HPLCを組み合わせて、さらに二次元PA化糖鎖マッピング、エキソグリコシダーゼ消化、イオンスプレータンデムマススペクトロメトリー(IS−MS/MS)(Perkin Elmer社製)により解析した。まず、細胞抽出液をアセトンで脱脂後、100℃、12時間ヒドラジン処理し、糖鎖部分を遊離させた。ヒドラジン分解物を、N−アセチル化し、Dowex 50X2およびDowex 1X2(それぞれ、代理店室町化学工業株式会社、The Dow Chemical Co.社製)により脱塩し、0.1NアンモニアによりSephadex G−25ゲルろ過カラム(1.8×180cm)(Pharmacia社製)を用いて分画した。ピリジルアミノ化(PA化)は先に述べた通りである。PA化糖鎖を、Jasco 821−FP Intelligent Spectrophotometer付きJasco880−PU HPLC装置(日本分光社製)により、Cosmosil 5C18−P及びAsahipak NH2P−50をカラムに用いて分離した。溶出位置を、自ら調製したか、あるいは購入した(Wako ChemicalsおよびTAKARA SHUZO)既知の標準品と比較した。
【0075】
N−アセチル−β−D−グルコサミニダーゼ(Diplococcus pneumoniae,Boehringer Mannheim)あるいはマンノシダーゼ(タチナタマメ、Sigma)を用いたグリコシダーゼ消化は、Kimura,Y.ら、Biosci.Biotech.Biochem.56(2)、215−222、1992で述べた方法と同様の条件下で約1nmolのPA化糖鎖について行った。β−ガラクトシダーゼ(Diplococcus pneumoniae,Boehringer Mannheim)とAspergillus saitoi由来のα−1,2マンノシダーゼ(東北大学吉田孝博士より分与)による消化については、50mM 酢酸ナトリウム緩衝液(pH5.5)中に1nmolのPA化糖鎖を200mU β−ガラクトシダーゼあるいは60μg α−1,2マンノシダーゼを37℃で加温した。酵素反応を沸騰させて停止し、消化物の一部をサイズ分画HPLCにより解析した。生じた消化物の分子量はイオンスプレーマススペクトロメトリー(IS−MS/MS)により解析し、そして/あるいはPalacpac,N.Q.ら、Biosci.Biotech.Biochem.63(1)35−39、1999およびKimuraら、Biosci.Biotech.Biochem.56(2)、215−222、1992に示されているように標準糖鎖と比較した。
【0076】
IS−MS/MS実験は、Perkin Elmer Sciex API−IIIにより行った。ポジティブモードで、イオンスプレーの電圧を4200Vにて行った。スキャンニングを0.5Da毎で行い、m/zを200から記録した。
【0077】
(GT6細胞の糖鎖の解析)
GT6細胞より調製したPA化糖鎖を精製し、逆相HPLCとサイズ分画HPLCを組み合わせて解析した。サイズ分画HPLCのもとで、10〜20分の位置のフラクションI(図11)には、N−結合型糖鎖は溶出しておらず、ヒドラジン分解による副産物を含む非吸着部分であることを示唆している。MS/MS解析では、PA−GlcNAcに相当する300というm/z値を持つフラグメントイオンを検出できなかった。同様に、50〜60分の位置のフラクションXIも、サイズ分画HPLCで溶出を示すピークを検出できなかったので、N−結合型糖鎖を含んでいないことがわかった。しかし、サイズ分画HPLC(図11)におけるフラクションIIからXまでの解析から、図12に示すA〜Qを含む全部で17のピークを回収および精製した。
【0078】
IS−MS/MS分析は、これらのピークのうち7つのピークがN−結合型糖鎖であることを示した。以下各ピークの分析結果を示す。
【0079】
オリゴ糖−A、−E、−H、−I、−M、−O、−P、及び−Q(図12)の溶出位置及び分子量は、PA化糖鎖標準品と対応せず、MS/MS解析ではPA−GlcNAc及びPA−GlcNAc2に相当する300及び503というm/z値を有していたが、N−結合型糖鎖一般的に認められるManGlcNA2(M1)或いはトリマンノースコア糖鎖Man3GlcNAc2(M3)に相当するフラグメントイオンを検出できなかった(データ未掲載)。他のピークのオリゴ糖−B,−D及び−Nも、300というm/z値を持つフラグメントイオンを検出できなかったので、N−結合型ではなかった。そこで残りの7つのN−結合型糖鎖について調べた。
【0080】
ピーク−C(m/z1637.5;モル存在比9.3%)、ピーク−F([M+2H]2+ m/z819.5,[M+H]+ m/z1639;モル存在比15.9%)、およびピーク−G(m/z1475.5 モル存在比19.5%)の溶出位置と分子量はそれぞれ、高マンノース型糖鎖Man7GlcNAC2(アイソマーM7A及びM7B)、およびMan6GlcNAc2(M6B)であった。タチナタマメ α−マンノシダーゼで消化すると、各N−結合型糖鎖は、サイズ分画HPLCによりManGlcNAc(M1)へと分解されていることを示した(データ未掲載)。消化物のIS−MS実験によりm/z値665.5というイオンは、M1の計算値664.66に対応していたことから、これらは対応する個々のPA化糖鎖標準品と構造的に同一であることを示している。
【0081】
ピーク−J(6.6%)は、分子量1121.5を有していたが、これはMan3Xyl1GlcNAc2−PA(M3X)の分子量計算値m/z1121.05と同じであった。989.5、827.5、665.5、503.3及び300に見られたフラグメントイオンの位置は、Man3Xyl1GlcNAc2−PAから逐次Xyl、Man、Man、Man及びGlcNAcがこの順序で遊離したことと矛盾しない。タチナタマメα−マンノシダーゼで処理すると、非還元末端のマンノース残基を除去でき、2次元マッピングはMan1Xyl1GlcNAc2−PAの溶出位置と同じであった(データ未掲載)。
【0082】
ピーク−K(13.2%)のIS−MS実験の解析結果は、このフラクションが2種のN−結合型糖鎖を含むことを示し、そのひとつは分子量が1314.0(1.4%)で他方が1354.5(11.8%)だった。このフラクションをさらに逆相HPLCに供し、精製し、更に解析した。分子量1314.0を有する糖鎖ピーク−K−1は、糖鎖標準品Man5GlcNAc2−PA(M5)と二次元マッピング及びm/z測定値が同一だった。さらに、タチナタマメα−マンノシダーゼで処理すると、その分解産物の溶出位置が2次元マッピングでM1と同じ位置に移動したことら、4つのマンノース残基が除去されたことを示した。
【0083】
(GT6細胞中のガラクトースが付加したN−結合型糖鎖)
m/z1354.5を有する糖鎖ピーク−K−2は、Gal1GlcNAc1Man3GlcNAc2−PA(GalGNM3)の予想される分子量m/z1354.3とよく一致していた。質量分析の結果から、親分子から派生したと考えられるフラグメントイオンはそれぞれ、m/z1193.5がGlcNAc1Man3GlcNAc2−PA、m/z989.5がMan3GlcNAc2−PA、m/z827.5がMan2GlcNAc2−PA、m/z665がManGlcNAc2−PA、m/z503がGlcNAc2−PA、m/z336がManGlcNAc、m/z300がGlcNAc−PAそしてm/z204がGlcNAcであった。推定されるN−結合型糖鎖構造から、GalGNM3の2種のアイソマーが考えられる(図13)。すなわち、Galβ4GlcNAcβ2Manα6(Manα3)Manβ4GlcNacβ4GlcNAc−PA及びManα6(Galβ4GlcNAcβ2Manα3)Manβ4GlcNAcβ4GlcNAc−PAである。精製したPA化糖鎖は、糖鎖標準品Manα6(Galβ4GlcNAcβ2Manα3)Manβ4GlcNAcβ4GlcNAc−PAと同一の逆相HPLCの溶出位置であった(図13のB)。
【0084】
糖鎖をエキソグリコシダーゼで処理し、そして糖鎖構造を確認した。D.pneumoniae β−ガラクトシダーゼはGalβ1−4GlcNAc結合特異的であるが、本酵素による消化物はGlcNAc1Man3GlcNAc2−PAとサイズ分画HPLCにて同一の位置に溶出した(図14A−II)。また、IS−MS/MS分析からm/z1192.0が得られた。これらの結果は、1つのガラクトース残基が、β1−4結合している非還元末端のGlcNAcから除去されたことを示している。さらにこの産物を、β1−2GlcNAc結合特異的なDiplococcus pneumoniae由来のN−アセチル−β−D−グルコサミニダーゼ(Yamashita,K.ら J.Biochem.93,135−147、1983)で消化すると、その消化物は標準品Man3GlcNAc2−PAとサイズ分画HPLCにて同じ位置に溶出した(図14A−III)。続いてタチナタマメα−マンノシダーゼで処理すると、標準品ManGlcNAc2−PAとサイズ分画HPLCにて同じ位置に溶出した(図14A−IV)。糖鎖構造を図15のK−2に示す。
【0085】
ピーク−L(35.5%)は、質量分析結果から[M+2H]2+が840、[M+H]+が1680.0であり、Gal1GlcNAc1Man5GlcNAc2−PA(GalGNM5)の予想される分子量m/z1678.55とよく一致していた。質量分析の結果から、親分子から派生したと考えられるフラグメントイオンはそれぞれ、m/z1313.5がMan5GlcNAc2−PA、m/z1152がMan4GlcNAc2−PA、m/z989.5がMan3GlcNAc2−PA、m/z827.5がMan2GlcNAc2−PA、m/z665がManGlcNAc2−PA、m/z503がGlcNAc2−PA、m/z336がManGlcNAc、m/z300がGlcNAc−PAそしてm/z204がGlcNAcであった。D. pneumoniae β−ガラクトシダーゼによる消化物はGlcNAc1Man5GlcNAc2−PAとサイズ分画HPLCにて同一の位置に溶出した(図14B−II)。この結果は、1つのガラクトース残基が、β1−4結合している非還元末端のGlcNAcに結合していたことを示している。ガラクトースが除去されたことを、IS−MS/MS分析により得られた分子量が[M+2H]2+が759、[M+H]+が1518.0であることにより確認した。親シグナルm/z1518.0であるGlcNAc1Man5GlcNAc2−PAから、フラグメントイオンm/z1314がMan5GlcNAc2−PA、m/z1152がMan4GlcNAc2−PA、m/z990がMan3GlcNAc2−PA、m/z827.5がMan2GlcNAc2−PA、m/z665.5がMan1GlcNAc2−PA、m/z503がGlcNAc2−PA、m/z300がGlcNAc−PAとが派生していた。GlcNAc1Man5GlcNAc2−PAをDiplococcus pneumoniae由来のN−アセチル−β−D−グルコサミニダーゼで消化すると、その消化物は標準品Man5GlcNAc2−PAとサイズ分画HPLCにて同じ位置に溶出した(図14B−III)。Aspergillus saitoi由来のα−1,2マンノシダーゼで処理しても溶出位置が移動しなかった(図14B−IV)。しかし、続いてタチナタマメ α−マンノシダーゼで処理すると、標準品Man1GlcNAc2−PAとサイズ分画HPLCにて同じ位置に溶出した(図14B−V)。このことは、非還元末端に存在する4つのマンノース残基が除去されたことを示している。これらの結果から、PA化糖鎖は、5つのいずれのマンノース残基も、α1−3結合したマンノース残基にα1−2結合していないことを示す。以上エキソグリコシダーゼ消化、2次元糖鎖マッピング、IS−MS/MS分析から導きだされる糖鎖構造は、GalGNM5で、図15のLに示す。
【0086】
図20は、GT6細胞株におけるN−結合グリカンの構造と各N−結合グリカンの比率に関する上記の結果を、同様に決定された野生型BY2細胞株におけるそれらとともに要約する。図20において、白抜き四角はGlcNAcを、白抜き丸はマンノースを、黒丸はガラクトースを、斜線をひいた四角はキシロースを、そして点をつけた丸はフコースをそれぞれ示す。
【0087】
GT6細胞株では、アイソマー類であるMan7−、Man6−およびMan5GlcNAc2が観察された。これらの高マンノース型オリゴサッカライドは、β1,4−ガラクトシルトランスフェラーゼ(GalT)の基質であるので、GlcNAcI、ManIおよびManII cDNAの導入は、オリゴサッカライドMan7−5GlcNAc2を、GalTの基質であり得るGlcNAcMan3GlcNAc2へより効率的に導き得る(図21)。
【0088】
GnT Iを欠くA.thaliana cglI変異体は、複合型N−グリカンを合成することができない(von Schaewen,A.,Sturm,A.,O’Neill、J.,およびChrispeels、MJ.,Plant Physiol.,1993 Aug;102(4):1109−1118、N−アセチルグルコサミニルトランスフェラーゼIを欠き、そしてゴルジ体で修飾される複合体N−結合グリカンを合成できない変異体Arabidopsis植物の単離)。このcglI変異体におけるヒトGnTでの相補性は、哺乳動物酵素が、植物N−グリコシル化経路に寄与し得たことを示した(Gomez、L.およびChrispeels,M.J.,Proc.Natl.Acad.Sci.USA 1994 3月 1;91(5)1829−1833、複合アスパラギン結合グリカンを欠くArabidopsis thalianaの、N−アセチルグルコサミニルトランスフェラーゼIをコードするヒトcDNAでの相補性)。さらに、A.thalianaから単離されたGnT I cDNAは、CHO Lec1細胞のN−アセチルグルコサミニルトランスフェラーゼI欠損を相補した(Bakker,H.,Lommen,A.,Jordi,W.,Stiekema,W.,およびBosch,D.,Biochem.Biophys.Res.Commun.,1999 8月 11;261(3):829−32、A.thaliana cDNAは、CHO Lec1細胞のN−アセチルグルコサミニルトランスフェラーゼI欠損を相補する)。ヒトManIおよびManIIをコードするcDNAが単離され、そして配列決定された(Bause,E.,Bieberich,E.,Rolfs,A.,Volker,C.およびSchmidt,B.,Eur J Biochem 1993 10月 15;217(2):535−40、ヒト腎臓からのMan9−マンノシダーゼの分子クローニングおよび一次構造;Tremblay,L.O.,Campbell,Dyke,N.およびHerscovics,A.,Glycobiology 1988 6月;8(6):585−95、N−グリカン成熟に関与する新規ヒトα1,2−マンノシダーゼ遺伝子の分子クローニング、染色体マッピングおよび組織特異的発現;およびMisago.M.,Liao,Y.F.,Kudo,S.,Eto,S.,Mattei,M.G.,Moremen,K.W.,Fukuda,M.N.,ヒトα−マンノシダーゼIIおよび従前には認識されていないα−マンノシダーゼIIxアイソザイムをコードするcDNAの分子クローニングおよび発現)。ヒトManIは、2つのアイソザイムManIAおよびManIBをもち、そしてアイソザイムのcDNAのヌクレオチド構造が示された(Bause,E.,ら、およびTremblay,L.O.,前述)。これらのcDNAを、BY細胞株中に形質転換することにより、ヒト型糖タンパク質を産生する効率的な細胞株が得られ得る。β1,4−ガラクトシルトランスフェラーゼ(GalT)は、UDP−ガラクトースをドナー基質として用い、そしてGlcNAc2Man3GlcNAc2をアクセプター基質として用いる。UDP−ガラクトースの効率的な供給はGalT酵素反応を増大し、そしてより多くのガラクトシル化オリゴサッカライドが産生される(図22)。
【0089】
(実施例7)二重形質転換体GT6−HRP細胞中のHRPの糖鎖の構造解析
GT6−HRP細胞またはコントロールのBY2−HRP細胞の7日間培養した培養液のそれぞれのホモゲナイズ物50gから得た粗細胞溶解液を、10mMリン酸ナトリウム緩衝液(pH6.0)で平衡化したCM Sepharose FFカラム(1×10cm)(Pharmacia社製)にかけた。カラムを洗浄後、溶出したペルオキシダーゼを403nmの吸光度で測定した。プールした画分を限外濾過(分子量カット:10,000、Advantec社製)により濃縮し、50mMのリン酸ナトリウム緩衝液(pH7.0)中で透析し、そして平衡化したベンズヒドロキサミン酸−アガロースアフィニティーカラム(1×10cm)(KemEn Tec,Denmark)にかけた。カラムを15倍容量の50mMリン酸ナトリウム緩衝液(pH7.0)で洗浄した後、同じ緩衝液中に調製した0.5Mのホウ酸で、結合したHRPを溶出した。得られたペルオキシダーゼ活性画分をプールし、透析および濃縮した。
【0090】
形質転換細胞GT6−HRP細胞およびBY2−HRP細胞のそれぞれから得た精製HRPを、1×10cmのRCA120−アガロースカラムにかけた。次いでカラムを、15倍容量の10mM酢酸アンモニウム(pH6.0)で洗浄した。結合タンパク質を、常法に従って溶出し、アッセイした。
【0091】
β1−4結合ガラクトースに特異的であるRCA120アフィニティークロマトグラフィーから溶出された精製HRPのレクチン染色では、レクチンRCA120が、形質転換細胞GT6−HRPで産生されたHRPのみを結合した。レクチンを競合するガラクトースとプレインキュベートすることによりレクチン結合は劇的に減少したので(図16bIII)、この結合は炭水化物特異的であった。D.pneumoniae β−ガラクトシダーゼで精製HRPを前処理しても、RCA120結合を阻害した。これらの結果は、RCAがHRP上のN結合型オリゴ糖鎖の末端β1−4結合位置にあるガラクトースに特異的に結合したことを示す。BY2−HRP細胞中にRCA結合糖タンパク質がないことは、この細胞がHRPグリカンの非還元末端にβ1−4結合ガラクトース残基を転移し得ないことを示す。
【0092】
RCA120を用いて精製したHRP由来のPA誘導体の逆相HPLC(Jasco821−FP Intelligent Spectrofluorometerを備えたJasco880−PU HPLC装置上、Cosmosil 5C18−PカラムまたはAsahipakNH2Pカラムを用いた)は、GT6−HRPより精製されたHRPタンパク質上の糖鎖が単一のピークを示した(図17)。一方、BY2−HRPより精製されたHRP画分からは、結合タンパク質または検出可能なピークは観察されなかった。GT6−HRPから得られたピークは、サイズ分画HPLC上で均一であった。二次元マッピング分析および標準糖鎖との同時クロマトグラフィーは、このオリゴサッカライドがGal1GlcNAc1Man5GlcNAc2−PAと一致することが判明した。この構造の確認は、連続的エクソグリコシダーゼ消化によりなされた。標準糖鎖としては、先に調製された糖鎖(Kimura,Yら、Biosci.Biotech.Biochem.56(2),215−222,1992)または市販(Wako Chemicals,OsakaおよびTAKARA SHUZO)の糖鎖を用いた。
【0093】
PA糖鎖のβ−ガラクトシダーゼ(D.pneumoniae)消化は、標準のGlcNAc1Man5GlcNAc2−PAの溶出位置と一致し、1つの非還元末端GlcNAcにβ1−4結合した1つのガラクトース残基が除去されたことを示した。D.pneumoniae由来のN−アセチル−β−D−グルコサミニダーゼでのこの消化物のさらなる消化は、Man5GlcNAc2−PAの溶出位置に等しい糖鎖を生じ、1つの非還元末端マンノース残基にβ1−2結合したGlcNAc残基を損失したことを示した。植物におけるN結合型プロセッシング経路に基づけば、除去されたGlcNAc残基は、β1−4マンノース残基に連結したα1−3マンノースにあると考えられる。GlcNAc残基の結合位置を確立するために、Man5GlcNAc2−PA(M5)を、Aspergillus saitoi由来α1−2マンノシダーゼとともにインキュベートした。予想されたように、溶出位置にシフトは見られず、M5が予想された構造Manα1−6(Manα1−3)Manα1−6(Manα1−3)Manβ1−4GlcNAcβ1−4GlcNAcを有することが確認された。糖鎖をタチナタマメα−マンノシダーゼを用いてさらに切りそろえると、公知のMan1GlcNAc2−PAの溶出位置になった。従って、この糖鎖構造は、Manα1−6(Manα1−3)Manα1−6(Galβ1−4GlcNAcβ1−2Manα1−3)Manβ1−4GlcNAcβ1−4GlcNAc(Gal1GlcNAc1Man5GlcNAc2)に対応する。以上の結果から、GT6細胞中に存在する糖鎖構造としては図15に示される通りであり、二重形質転換体GT6−HRP細胞由来のHRPタンパク質の有する糖鎖構造は、Manα1−6(Manα1−3)Manα1−6(Galβ1−4GlcNAcβ1−2Manα1−3)Manβ1−4GlcNAcβ1−4GlcNAc(Gal1GlcNAc1Man5GlcNAc2)であることが導き出される。
【0094】
同様に、形質転換細胞GT6−HRP由来のHRPのガラクトシル化N−グリカンは、植物のN−グリカンに特徴的なβ1,2キシロース残基と特異的に反応することが示されている抗血清と反応せず、植物の複合型グリカン中の抗原性を引き起こすことが示されている糖残基(キシロース残基)(Garcia−Casado,G.ら,Glycobiology 6(4):471−477,1996)の1つが存在しないことを示した(図18)。
【0095】
(発明の効果)
本発明により、ヒト型糖鎖をもつ糖タンパク質の生産方法;非還元末端アセチルグルコサミン残基へのガラクトース残基の転移反応を行い得る糖鎖付加機構を備え、この糖鎖付加機構が、コア糖鎖および外部糖鎖を含む糖鎖であって、コア糖鎖が複数のマンノースおよびアセチルグルコサミンから本質的になり、外部糖鎖が非還元末端ガラクトースを含む末端糖鎖部分を含む糖鎖、を付加する、植物細胞;およびこの方法によって得られたヒト型の糖鎖をもつ糖タンパク質が提供される。本発明のヒト型の糖鎖をもつ糖タンパク質は、糖付加がヒト型であるために、抗原性を有さない。それゆえ、ヒトを含む動物への投与のために有用である。
【特許請求の範囲】
【請求項1】
糖鎖をもつ糖タンパク質の生産方法であって、哺乳動物のグリコシルトランスフェラーゼの遺伝子および糖タンパク質をコードする遺伝子を植物細胞に導入することによって形質転換植物細胞を得る工程であって、ここで、該糖タンパク質が該植物細胞には本来存在せず、該酵素がグリコシル残基を該糖タンパク質に転移する前記工程、および得られた形質転換植物細胞を培養する工程を含む前記生産方法。
【請求項2】
前記哺乳動物のグリコシルトランスフェラーゼが、哺乳動物のβ1,4−ガラクトシルトランスフェラーゼであり、前記グリコシル残基がガラクトシル残基である、請求項1に記載の方法。
【請求項3】
前記糖タンパク質がフコースおよびキシロースを含まない、請求項2に記載の方法。
【請求項4】
前記糖タンパク質の糖鎖が、コア糖鎖および外部糖鎖を含み、ここで、該コア糖鎖が複数のマンノースおよびアセチルグルコサミン残基を含み、そして該外部糖鎖が、非還元末端ガラクトースをもつ末端糖部分を含む、請求項2に記載の方法。
【請求項5】
前記外部糖鎖が直鎖状構造をもつ、請求項4に記載の方法。
【請求項6】
前記外部糖鎖が分岐状構造をもつ、請求項4に記載の方法。
【請求項7】
前記分岐糖鎖部分が、モノ、バイ、トリ、またはテトラ構造をもつ、請求項6に記載の方法。
【請求項8】
前記哺乳動物のβ1,4−ガラクトシルトランスフェラーゼが、ヒトβ1,4−ガラクトシルトランスフェラーゼである、請求項2に記載の方法。
【請求項9】
第2の哺乳動物のグリコシルトランスフェラーゼ酵素をコードする遺伝子を導入することをさらに含む、請求項1に記載の方法。
【請求項10】
前記第2の酵素が、N−アセチルグリコサミニルトランスフェラーゼI(GlcNAcI)である、請求項9に記載の方法。
【請求項11】
植物細胞に本来存在しない前記糖タンパク質が、西洋ワサビペルオキシダーゼ、キナーゼ、グルコセレブロシダーゼ(glucocerebrosidase)、α−ガラクトシダーゼ、組織型プラスミノーゲン活性化因子(TPA)、3−ヒドロキシ−3−メチルグルタリル補酵素A(HMG−CoA)レダクターゼ、エンケファリン、インターフェロン−アルファ、顆粒球マクロファージコロニー刺激因子(GM−CSF)、顆粒球コロニー刺激因子(G−CSF)、絨毛性性腺刺激ホルモン、インターロイキン−2、インターフェロン−ベータ、インターフェロン−ガンマ、エリスロポイエチン、血管内皮細胞増殖因子(vascular endothelial growth factor)、ヒト絨毛性ゴナドトロピン(HCG)、黄体形成ホルモン(LH)、甲状腺刺激ホルモン(TSH)、プロラクチン、卵胞刺激ホルモン、IgG抗体、および一本鎖可変領域抗体フラグメント(scFV)からなる群から選択される、請求項1に記載の方法。
【請求項12】
哺乳動物のグリコシルトランスフェラーゼをコードする遺伝子、および外因性糖タンパク質をコードする遺伝子を含む形質転換植物細胞であって、ここで、該外因性糖タンパク質が発現され、発現した糖タンパク質がヒト型糖鎖を含む前記形質転換植物細胞。
【請求項13】
前記哺乳動物のグリコシルトランスフェラーゼが、ガラクトース残基を非還元末端アセチルグルコサミン残基を転移する哺乳動物のβ1,4−ガラクトシルトランスフェラーゼである、請求項12に記載の形質転換植物細胞。
【請求項14】
前記ヒト型糖鎖をもつ糖タンパク質が、コア糖鎖および外部糖鎖を含み、ここで、該コア糖鎖が複数のマンノースおよびアセチルグルコサミンを含み、そして該外部糖鎖が、非還元末端ガラクトースをもつ末端糖鎖部分を含む、請求項13に記載の形質転換植物細胞。
【請求項15】
前記糖タンパク質が、コア糖鎖、外部糖鎖および末端糖鎖の1以上に連結したフコース及びキシロースを含まない、請求項13に記載の形質転換植物細胞。
【請求項16】
前記哺乳動物のβ1,4−グリコシルトランスフェラーゼがヒトβ1,4−ガラクトシルトランスフェラーゼであり、前記外因性糖タンパク質がホルモン、サイトカイン、抗体、ワクチン、レセプターまたは血清タンパク質である、請求項13に記載の形質転換植物細胞。
【請求項17】
前記第2の哺乳動物のグリコシルトランスフェラーゼ酵素をコードする遺伝子をさらに含む、請求項12に記載の形質転換植物細胞。
【請求項18】
前記第2の酵素が、N−アセチルグリコサミニルトランスフェラーゼI(GlcNAcI)である、請求項17に記載の形質転換植物細胞。
【請求項19】
請求項12に記載の植物細胞から再生される形質転換植物。
【請求項20】
哺乳動物のグリコシルトランスフェラーゼをコードする遺伝子、および外因性糖タンパク質をコードする遺伝子を含む形質転換植物であって、該外因性糖タンパク質が発現され、発現された糖タンパク質がヒト型糖鎖を含む前記形質転換植物又はその一部分。
【請求項21】
前記哺乳動物のグリコシルトランスフェラーゼが、ガラクトース残基を非還元末端アセチルグルコサミン残基に転移する哺乳動物のβ1,4−ガラクトシルトランスフェラーゼである、請求項20に記載の形質転換植物。
【請求項22】
前記ヒト型糖鎖をもつ糖タンパク質が、コア糖鎖および外部糖鎖を含み、ここで、該コア糖鎖が複数のマンノースおよびアセチルグルコサミンを含み、そして該外部糖鎖が、非還元末端ガラクトースをもつ末端糖鎖部分を含む、請求項21に記載の形質転換植物。
【請求項23】
前記生成された糖タンパク質が、コア糖鎖、外部糖鎖および末端糖鎖の1以上に連結したフコース及びキシロースを含まない、請求項21に記載の形質転換植物。
【請求項24】
前記哺乳動物のβ1,4−グリコシルトランスフェラーゼがヒトβ1,4−ガラクトシルトランスフェラーゼであり、前記外因性糖タンパク質がホルモン、サイトカイン、抗体、ワクチン、レセプターまたは血清タンパク質である、請求項21に記載の形質転換植物。
【請求項25】
第2の哺乳動物のグリコシルトランスフェラーゼ酵素をコードする遺伝子をさらに含む、請求項20に記載の形質転換植物。
【請求項26】
前記第2の酵素が、N−アセチルグリコサミニルトランスフェラーゼI(GlcNAcI)である、請求項25に記載の形質転換植物。
【請求項1】
糖鎖をもつ糖タンパク質の生産方法であって、哺乳動物のグリコシルトランスフェラーゼの遺伝子および糖タンパク質をコードする遺伝子を植物細胞に導入することによって形質転換植物細胞を得る工程であって、ここで、該糖タンパク質が該植物細胞には本来存在せず、該酵素がグリコシル残基を該糖タンパク質に転移する前記工程、および得られた形質転換植物細胞を培養する工程を含む前記生産方法。
【請求項2】
前記哺乳動物のグリコシルトランスフェラーゼが、哺乳動物のβ1,4−ガラクトシルトランスフェラーゼであり、前記グリコシル残基がガラクトシル残基である、請求項1に記載の方法。
【請求項3】
前記糖タンパク質がフコースおよびキシロースを含まない、請求項2に記載の方法。
【請求項4】
前記糖タンパク質の糖鎖が、コア糖鎖および外部糖鎖を含み、ここで、該コア糖鎖が複数のマンノースおよびアセチルグルコサミン残基を含み、そして該外部糖鎖が、非還元末端ガラクトースをもつ末端糖部分を含む、請求項2に記載の方法。
【請求項5】
前記外部糖鎖が直鎖状構造をもつ、請求項4に記載の方法。
【請求項6】
前記外部糖鎖が分岐状構造をもつ、請求項4に記載の方法。
【請求項7】
前記分岐糖鎖部分が、モノ、バイ、トリ、またはテトラ構造をもつ、請求項6に記載の方法。
【請求項8】
前記哺乳動物のβ1,4−ガラクトシルトランスフェラーゼが、ヒトβ1,4−ガラクトシルトランスフェラーゼである、請求項2に記載の方法。
【請求項9】
第2の哺乳動物のグリコシルトランスフェラーゼ酵素をコードする遺伝子を導入することをさらに含む、請求項1に記載の方法。
【請求項10】
前記第2の酵素が、N−アセチルグリコサミニルトランスフェラーゼI(GlcNAcI)である、請求項9に記載の方法。
【請求項11】
植物細胞に本来存在しない前記糖タンパク質が、西洋ワサビペルオキシダーゼ、キナーゼ、グルコセレブロシダーゼ(glucocerebrosidase)、α−ガラクトシダーゼ、組織型プラスミノーゲン活性化因子(TPA)、3−ヒドロキシ−3−メチルグルタリル補酵素A(HMG−CoA)レダクターゼ、エンケファリン、インターフェロン−アルファ、顆粒球マクロファージコロニー刺激因子(GM−CSF)、顆粒球コロニー刺激因子(G−CSF)、絨毛性性腺刺激ホルモン、インターロイキン−2、インターフェロン−ベータ、インターフェロン−ガンマ、エリスロポイエチン、血管内皮細胞増殖因子(vascular endothelial growth factor)、ヒト絨毛性ゴナドトロピン(HCG)、黄体形成ホルモン(LH)、甲状腺刺激ホルモン(TSH)、プロラクチン、卵胞刺激ホルモン、IgG抗体、および一本鎖可変領域抗体フラグメント(scFV)からなる群から選択される、請求項1に記載の方法。
【請求項12】
哺乳動物のグリコシルトランスフェラーゼをコードする遺伝子、および外因性糖タンパク質をコードする遺伝子を含む形質転換植物細胞であって、ここで、該外因性糖タンパク質が発現され、発現した糖タンパク質がヒト型糖鎖を含む前記形質転換植物細胞。
【請求項13】
前記哺乳動物のグリコシルトランスフェラーゼが、ガラクトース残基を非還元末端アセチルグルコサミン残基を転移する哺乳動物のβ1,4−ガラクトシルトランスフェラーゼである、請求項12に記載の形質転換植物細胞。
【請求項14】
前記ヒト型糖鎖をもつ糖タンパク質が、コア糖鎖および外部糖鎖を含み、ここで、該コア糖鎖が複数のマンノースおよびアセチルグルコサミンを含み、そして該外部糖鎖が、非還元末端ガラクトースをもつ末端糖鎖部分を含む、請求項13に記載の形質転換植物細胞。
【請求項15】
前記糖タンパク質が、コア糖鎖、外部糖鎖および末端糖鎖の1以上に連結したフコース及びキシロースを含まない、請求項13に記載の形質転換植物細胞。
【請求項16】
前記哺乳動物のβ1,4−グリコシルトランスフェラーゼがヒトβ1,4−ガラクトシルトランスフェラーゼであり、前記外因性糖タンパク質がホルモン、サイトカイン、抗体、ワクチン、レセプターまたは血清タンパク質である、請求項13に記載の形質転換植物細胞。
【請求項17】
前記第2の哺乳動物のグリコシルトランスフェラーゼ酵素をコードする遺伝子をさらに含む、請求項12に記載の形質転換植物細胞。
【請求項18】
前記第2の酵素が、N−アセチルグリコサミニルトランスフェラーゼI(GlcNAcI)である、請求項17に記載の形質転換植物細胞。
【請求項19】
請求項12に記載の植物細胞から再生される形質転換植物。
【請求項20】
哺乳動物のグリコシルトランスフェラーゼをコードする遺伝子、および外因性糖タンパク質をコードする遺伝子を含む形質転換植物であって、該外因性糖タンパク質が発現され、発現された糖タンパク質がヒト型糖鎖を含む前記形質転換植物又はその一部分。
【請求項21】
前記哺乳動物のグリコシルトランスフェラーゼが、ガラクトース残基を非還元末端アセチルグルコサミン残基に転移する哺乳動物のβ1,4−ガラクトシルトランスフェラーゼである、請求項20に記載の形質転換植物。
【請求項22】
前記ヒト型糖鎖をもつ糖タンパク質が、コア糖鎖および外部糖鎖を含み、ここで、該コア糖鎖が複数のマンノースおよびアセチルグルコサミンを含み、そして該外部糖鎖が、非還元末端ガラクトースをもつ末端糖鎖部分を含む、請求項21に記載の形質転換植物。
【請求項23】
前記生成された糖タンパク質が、コア糖鎖、外部糖鎖および末端糖鎖の1以上に連結したフコース及びキシロースを含まない、請求項21に記載の形質転換植物。
【請求項24】
前記哺乳動物のβ1,4−グリコシルトランスフェラーゼがヒトβ1,4−ガラクトシルトランスフェラーゼであり、前記外因性糖タンパク質がホルモン、サイトカイン、抗体、ワクチン、レセプターまたは血清タンパク質である、請求項21に記載の形質転換植物。
【請求項25】
第2の哺乳動物のグリコシルトランスフェラーゼ酵素をコードする遺伝子をさらに含む、請求項20に記載の形質転換植物。
【請求項26】
前記第2の酵素が、N−アセチルグリコサミニルトランスフェラーゼI(GlcNAcI)である、請求項25に記載の形質転換植物。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】

【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【公開番号】特開2012−223198(P2012−223198A)
【公開日】平成24年11月15日(2012.11.15)
【国際特許分類】
【出願番号】特願2012−171688(P2012−171688)
【出願日】平成24年8月2日(2012.8.2)
【分割の表示】特願2008−262161(P2008−262161)の分割
【原出願日】平成11年12月8日(1999.12.8)
【出願人】(510302021)フィトン ホールディングス,リミティド ライアビリティ カンパニー (1)
【Fターム(参考)】
【公開日】平成24年11月15日(2012.11.15)
【国際特許分類】
【出願日】平成24年8月2日(2012.8.2)
【分割の表示】特願2008−262161(P2008−262161)の分割
【原出願日】平成11年12月8日(1999.12.8)
【出願人】(510302021)フィトン ホールディングス,リミティド ライアビリティ カンパニー (1)
【Fターム(参考)】
[ Back to top ]