
ペプチドの新規な合成方法
【課題】 本発明は、ペプチド同士を直接結合させることにより新規なペプチドをブロック単位で合成する方法を提供することを課題とする。
【解決手段】 シアノシステイン残基を有するペプチド鎖「R1-CO-NH-C(CH2-SCN)-CO-NH-R2」にアミノ末端がフリーな別のペプチド鎖「NH2-R3」を作用させ、ペプチド鎖内のアミノ基にペプチド結合上のカルボニル炭素を求核攻撃させることにより、ペプチド間の結合反応を生じさせ、「R1-CO-NH-R3」の配列を持つペプチド鎖を合成した。
【解決手段】 シアノシステイン残基を有するペプチド鎖「R1-CO-NH-C(CH2-SCN)-CO-NH-R2」にアミノ末端がフリーな別のペプチド鎖「NH2-R3」を作用させ、ペプチド鎖内のアミノ基にペプチド結合上のカルボニル炭素を求核攻撃させることにより、ペプチド間の結合反応を生じさせ、「R1-CO-NH-R3」の配列を持つペプチド鎖を合成した。
【発明の詳細な説明】
【0001】
【発明の属する技術分野】本発明は、ペプチド工学の分野、特に、ペプチドの合成の分野に属する。
【0002】
【従来の技術】近年のペプチド工学の進歩に伴い、これまでに開発された純粋化学合成技術を駆使して最小活性単位ペプチドの迅速スクリーニング等により機能単位の部分ペプチドを明らかにする手法が開発されている。例えば、ランダムペプチドライブラリーと称される膨大な多様性を持つペプチドの一群を構築して、この中から有用な機能を有するペプチドを見つけだすことが行われるようになった(K.Janda,Proc.Natl.Acad.Sci.USA. 91, 10779-10785(1994))。
【0003】しかしながら、それらの機能単位のペプチドを組み合わせて、さらに高度の機能のペプチドの合成を試みる場合において、従来の手法を用いる場合、次のような問題点があった。即ち、従来の化学合成法では、その材料として保護アミノ酸を用いる必要があり、一旦、化学合成し、脱保護精製して得られたペプチドは、もはや化学合成の材料として用いることはできなかった。このため、機能単位のペプチドを組み合わせて、さらに高度の機能のペプチドを合成しようとする場合は、もはや保護基をもたないペプチドは化学合成の材料として利用できないという欠点があった。精製して得られたペプチドを原料として、従来の化学合成で行われているような保護を行うことなく、ペプチド同士を結合できれば、ペプチド工学の分野において画期的な技術となる。このため、ペプチドの末端どうしを結合させる技術の開発が試みられるようになった。例えば、Tamら(C.F.Liu, C.Rao, J.P.Tam, J.American Chemical Society, 118, 307(1996))は、一般に化学反応の進みにくいペプチド間の反応を、まずアルデヒドとシステイン間にチオエステル結合中間体を形成させ(キャプチャー反応(Capture反応))、その後近接効果に基づいたアリルリアレンジメント反応(Aryl rearrangement反応)によりアミド結合生成に導いている。しかしながら、この方法により生成される結合部位は天然のペプチド結合と異なり、5員環の環状構造を持つことに難点がある。また同様な方法がいくつか開発されているが、いずれも結合により生じた構造はいわゆるペプチド結合、「-CO-NH-」、とは全く異なった構造となっている。さらに、その反応も複雑である(三原久和、化学、51巻6号、396ページ)。このように、現在まで、ペプチド同士を組み合わせて、新規なペプチドを合成する効果的な方法は皆無に等しい。
【0004】
【発明が解決しようとする課題】本発明は、ペプチド同士を直接結合し、新規なペプチドをブロック単位で合成することを可能にする方法を提供することを課題とする。
【0005】
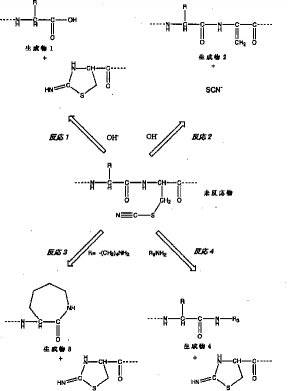
【課題を解決するための手段】本発明者等は、タンパク質中のシステイン残基のスルフヒドリル基を化学修飾することによりシアノシステイン残基に転換し、シアノシステイン残基を介したペプチド鎖切断の反応のメカニズムを検討し、図1に示される反応が関与することを明らかにした。
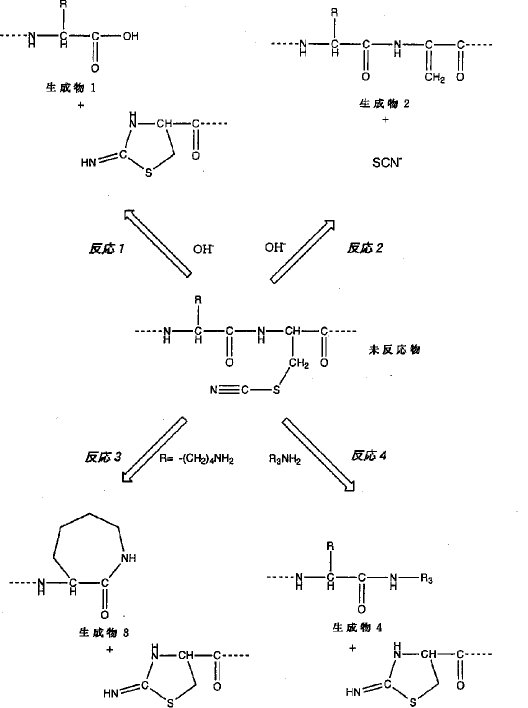
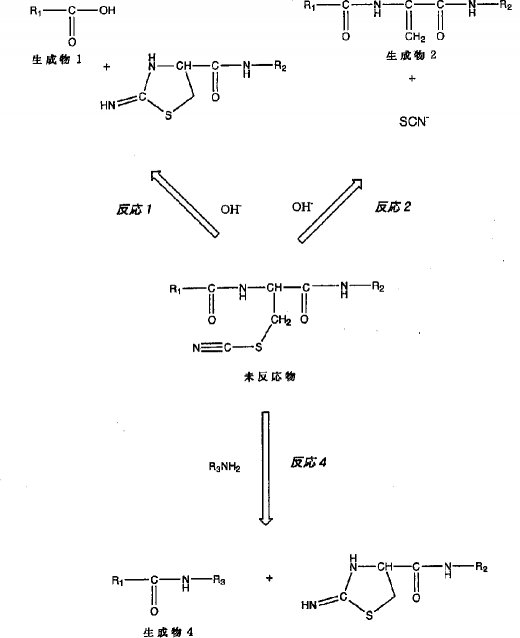
【0006】図1中、反応1は、ヤコブソン(Jacobson)ら(G.R.Jacobson, M.H.Schaffer, G.R.Stark, T.C.Vanaman, J.Biological Chemistry, 248, 6583-6591(1973)に記載)により報告された反応で、水酸基がシアノシステイン残基のN末端側に隣接するアミノ酸に由来するカルボニル炭素を求核的に攻撃することにより起こるペプチド鎖の切断反応であり、反応2は、水酸基が酸・塩基触媒として働くことにより、チオシアノ基が脱離するβ脱離反応で、シアノシステイン残基がデヒドロアラニンに転換する反応である(Y.Degani, A.Patchornik, Biochemistry, 13, 1-11(1974)に記載)。反応3は、発明者らが、リジン−シアノシステインの配列を有するペプチドを開裂させた時に得られた切断生成物を詳細に解析した結果見出した反応である。この反応3においては、カルボキシ末端がラクタムを含む7員環構造となることから、反応3はリジンのεアミノ基がカルボニル炭素を求核的に攻撃した結果起こったものと考えられる。本発明者等は、分子間でも、一級アミンがシアノシステイン残基のN末端側に隣接するアミノ酸に由来するカルボニル炭素を求核的に攻撃することができるはずであると考えた(図4)。即ち、本発明者等は、反応4によってどのような一級アミン誘導体でも、シアノシステイン残基の前までの配列に結合させることができ、且つ、生じる結合は、「-CO-NH-」、即ちペプチド結合であるという着想を得た。そして、本発明者等は、図1の反応4が実際起こることを証明し、この機構に基づき2つのペプチド鎖を容易に結合させることができることを示し、本発明を完成した。具体的には、図2の反応4に示されるように、シアノシステイン残基を有するペプチド鎖「R1-CO-NH-C(CH2-SCN)-CO-NH-R2」にアミノ末端がフリーな別のペプチド鎖「NH2-R3」を作用させ、ペプチド鎖内のアミノ基にペプチド結合上のカルボニル炭素を求核攻撃させることにより、ペプチド間の結合反応を生じさせ、「R1-CO-NH-R3」の配列を持つペプチド鎖を合成できること示し、本発明を完成した。
【0007】すなわち、本発明は、一般式(1) R1-CO-NH-C(CH2-SCN)-CO-NH-R2で示されるシアノシステイン残基を有するペプチド性化合物に、一般式(2) NH2-R3で示される一級アミンを有する化合物を作用させることにより、一般式(3) R1-CO-NH-R3で示される化合物を生成させる方法〔式中、R1、R2、R3は、任意のアミノ酸配列を表す〕に関する。
【0008】本発明に係る反応の本質からして、ここで、R1、R2、及びR3の化学的形態は限定されないことは明白である。従って、本発明は、R1、R2、及びR3としては、非天然アミノ鎖を含むペプチド鎖及びその誘導体、アルキル鎖及びその誘導体、糖鎖及びその誘導体、脂肪酸及びその誘導体、ポリヌクレオチド及びその誘導体、ポリデオキシヌクレオチド及びその誘導体、ビタミン類及びその誘導体、各種抗生物質、などが含まれる他幅広い化学基が利用可能である。
【0009】
【発明の実施の形態】本発明によって、図2の反応式4のごとく、前記一般式(1)「R1-CO-NH-C(CH2-SCN)-CO-NH-R2」で示される化合物に、前記一般式(2)「NH2-R3」で示される化合物を反応させることにより、前記一般式(3)「R1-CO-NH-R3」で示される化合物を生成させることができる。ここで、一般式(1)で示される化合物は、一般式(4) R1-CO-NH-C(CH2-SH)-CO-NH-R2、即ち、R1とR2の間にシステイン残基を有する化合物のシステインのスルフヒドリル基を選択的にシアノ化することにより生成することができる。
【0010】シアノ化試薬としては、通常、2-ニトロ-5-チオシアノ-安息香酸(2-nitro-5-thiocyanobenzoic acid (NTCB))(Y.Degani, A.Patchornik, Biochemistry, 13, 1-11(1974)に記載)を用いる方法が簡便である。シアノ化の手法によって、本発明が制限を受けないことは明白である。NTCBは市販のものをそのまま用いることができる。NTCBを用いたシステインのスルフヒドリル基のシアノ化は、pH7〜9の間で効率よく行うことができ、且つ、遊離するが、チオニトロ安息香酸(thionitrobenzoate)の412nmの吸光度の増加(分子吸光係数=13,600)でシアノ化の反応効率を調べることができる。また、システインのスルフヒドリル基のシアノ化は、文献(J.Wood, & N. Catsimpoolas, J. biological Chemistry, 233, 2887(1963))記載の方法に従い, スルフヒドリル基のシアンによる直接的酸化で行うこともできる。従って、シアノ化の手法によって、本発明が制限を受けないことは明白である。
【0011】一般式(1)の化合物「R1-CO-NH-C(CH2-SCN)-CO-NH-R2」への、一般式(2)の化合物「NH2-R3」の反応は、弱アルカリ条件下(pH8〜10)に、室温で行うことができる。実施例1においては、「NH2-R3」としてグリシンアミドを用いた例を示す。この場合、図1R>1に示される反応1及び反応2が競争反応となるが、ペプチド結合生成反応が最も効率良く進むことが示されている。実施例2においては、pH10の条件を用いて、「NH2-R3」として、グリシン-L-アラニン-L-アラニン-L-チロシン-L-アラニンを用いた結果を示しているが、ペプチド結合反応の生成物が主反応生成物であることが示されている。この結果は、図1の反応4が実際に起こることを証明しており、さらに、この機構に基づき、2つのペプチド鎖を容易に結合させることができたことを実証している。しかしながら、本発明のペプチド鎖結合反応は、実施例で示した結合反応条件には、限定されない。即ち、図1の反応4は、反応の原料である一般式(1)の化合物「R1-CO-NH-C(CH2-SCN)-CO-NH-R2」及び一般式(2)の化合物「NH2-R3」が溶解し互いに溶液中で混じりあう条件でありさえすれば、進行する。原料を溶解する溶媒としては、水溶性の溶媒である、水、メタノール、エタノールなどのアルコール類の他ペプチドの溶解に用いられる、ジメチルホルムアミド(DMF)、ジメチルスルホキサイド(DMSO)などが可能であるが、本発明は、用いる溶媒によっては限定されないことは明白である。反応温度は、室温で高い反応効率が得られるが、用いる溶媒が、凍結もしくは沸騰しない温度範囲であれば問題なく用いることができる。
【0012】反応の追跡は、高速液体クロマトグラフィーを連結した質量分析装置を用いて、生成物を質量数で同定・帰属し、定量することにより行うことができる。実施例における生成物の同定・帰属・定量は、「LC10A型高速液体クロマトグラフィー」(島津製作所製)を連結した「PE Sciex API III質量分析装置」(パーキンエルマー社製)を用いて行ったが、生成物を正確に同定・帰属・定量できる方法であればどのような方法で行ってよく、本発明が反応の追跡方法に制限されることはないことは明らかである。
【0013】
【実施例】
[実施例1] N-アセチル-L-アラニン-L-アラニン-グリシン-(S-シアノ)L-システイン-L-アラニン(以下、「acY-A-A-G-cC-A」と略す。質量=622)とグリシンアミド(G-NH2)との結合反応ペプチド内のシアノシステインとそのN末端側に隣接するアミノ酸残基との間のペプチド結合内に存在するカルボニル炭素に対して、アミノ基による分子間求核攻撃が可能であると考えられた。そこで、ペプチドとして「acY-A-A-G-cC-A」を用い、アミノ基を有する分子としてグリシンアミドを用いて、反応を行った。具体的には、0.05Mリン酸と0.1Mホウ酸からなる緩衝液(pHは、7〜10まで変動させた)に、終濃度1mg/mlとなるように 「acY-A-A-G-cC-A」を加え、さらに終濃度10mg/mlとなるようにグリシンアミド(G-NH2)を加え、室温で3時間反応させた。なお、本実験の対照としてグリシンアミドの非存在下で反応を行った。本反応の場合、シアノシステインのN末端側に隣接するアミノ酸がグリシンであるため、図1の反応3は、起こり得ない。従って、本実施例において起こりうる反応経路は、図2で示される反応1、反応2、及び反応4である。また、それぞれの反応生成物である、「R1-COOH」,「R1-CO-NH-(C=CH2)-CO-NH-R2」, 及び「R1-CO-NH-R3」のそれぞれの質量数は、423、563、及び480である。
【0014】本実験におけるグリシンアミド存在下での結果を表1に、非存在下での結果を表2にそれぞれ示す。
【0015】
【表1】
──────────────────────────────────── 反応生成物の割合(グリシンアミドの存在下)
──────────────────────────────────── pH acY-A-A-G-cC-A 反応1の 反応2の 反応4の (未反応物) 生成物1 生成物2 生成物4(質量数 622 423 563 480 )──────────────────────────────────── 7 〜100(%) 〜0(%) 〜0(%) 〜0(%)
8 96.8 0 1.2 2.0 9 65.2 0 4.7 14.1 10 32.1 0.5 5.9 26.4────────────────────────────────────
【0016】
【表2】
──────────────────────────────────── 反応生成物の割合(グリシンアミドの非存在下)
──────────────────────────────────── pH acY-A-A-G-cC-A 反応1の 反応2の 反応4の (未反応物) 生成物1 生成物2 生成物4(質量数 622 423 563 480 )──────────────────────────────────── 7 〜100(%) 〜0(%) 〜0(%) 〜0(%)
8 89.6 9.2 1.2 0 9 65.2 32.0 2.8 0 10 32.1 53.0 14.9 0 ────────────────────────────────────表2から明らかなように、グリシンアミドの非存在下では、塩基性条件における水酸化物イオンが、ペプチド結合上のカルボニル炭素に対して求核攻撃を行う、反応1が主反応となって、ペプチドの切断が起こり、「acY-A-A-G」(質量数=423)と2-イミノチアゾリジン-4-カルボキシル-アラニン(2-iminothiazolidine-4-carboxylyl-alanine)が生成した。また、水酸化物イオンが塩基として作用しβ-脱離反応が起こる結果、ペプチドの切断が生じない反応2が副反応となり、N-アセチル-L-アラニン-L-アラニン-グリシン-L-デヒドロアラニン−L-アラニン(以下、「acY-A-A-G-dA-A」と略す。質量=563)が生成した。
【0017】一方、表1から明らかなように、グリシンアミド存在下では、グリシンアミド中のアミノ基が、ペプチド結合上のカルボニル炭素へ求核攻撃を行う反応4が主反応となって、この結果、ペプチドの切断と共に新たなペプチドの合成が引き起こされ、N-アセチル-L-アラニン-L-アラニン-グリシン-グリシンアミド(以下、「acY-A-A-G-G−NH2」と称する。質量数=480)が生成した。特に、高pH条件下では、主反応である反応4が促進されると共に、グリシンアミド非存在下の場合と比較して、副反応である反応2が阻害された。
【0018】[実施例2] N-アセチル-L-アラニン-L-アラニン-グリシン-(S-シアノ)L-システイン-L-アラニン(acY-A-A-G-cC-A)とグリシン-L-アラニン-L-アラニン-L-チロシン-L-アラニン(以下、「G-A-A-Y-A」と称する)とのペプチド結合反応実施例1において、グリシンアミド内のアミノ基による、ペプチド結合上のカルボニル炭素に対しての求核攻撃が可能であることが証明された。そこで、次に、グリシンアミドのような単純なアミノ酸誘導体に代えて、ペプチドである「G-A-A-Y-A」を用いて、同様の反応を行った。具体的には、0.05Mリン酸と0.1Mホウ酸からなる緩衝液に、終濃度1mg/mlとなるように 「acY-A-A-G-cC-A」を、終濃度10mg/mlとなるように「G-A-A-Y-A」を加え、室温で3時間反応させた。なお、緩衝液のpHは、実施例1で最もペプチド結合反応が促進されたpH10とした。この結果を表3に示す。
【0019】
【表3】
──────────────────────────────────── 反応生成物の割合──────────────────────────────────── acY-A-A-G-cC-A 反応1の 反応2の 反応4の (未反応物) 生成物1 生成物2 生成物4(質量数 622 423 563 857 )──────────────────────────────────── 60.1(%) 5.8(%) 12.4(%) 21.7(%) ────────────────────────────────────表3から明らかなように、グリシンアミドに代えて、ペプチドである「G-A-A-Y-A」を用いた場合でも、ペプチド内のグリシン残基のアミノ基による求核攻撃(反応4)により、新たなペプチド、N-アセチル-L-アラニン-L-アラニン-グリシン-グリシン-L-アラニン-L-アラニン-L-チロシン-L-アラニン(「acY-A-A-G-G-A-A-Y-A]、質量数=857)が生成した。
【0020】
【発明の効果】本発明により、保護基を用いずに、簡易かつ迅速にペプチドの合成を行う画期的な技術が提供された。これにより、機能単位のペプチドを組み合わせて、さらに高度の機能のペプチドを合成するなど、ペプチド合成の分野を中心に幅広い利用が期待される。
【図面の簡単な説明】
【図1】シアノシステイン残基を介したペプチド鎖切断に関与する反応の模式図である。
【図2】シアノシステイン残基を介したペプチド鎖切断に関与する反応のうち本発明において関与している反応の模式図である。
【0001】
【発明の属する技術分野】本発明は、ペプチド工学の分野、特に、ペプチドの合成の分野に属する。
【0002】
【従来の技術】近年のペプチド工学の進歩に伴い、これまでに開発された純粋化学合成技術を駆使して最小活性単位ペプチドの迅速スクリーニング等により機能単位の部分ペプチドを明らかにする手法が開発されている。例えば、ランダムペプチドライブラリーと称される膨大な多様性を持つペプチドの一群を構築して、この中から有用な機能を有するペプチドを見つけだすことが行われるようになった(K.Janda,Proc.Natl.Acad.Sci.USA. 91, 10779-10785(1994))。
【0003】しかしながら、それらの機能単位のペプチドを組み合わせて、さらに高度の機能のペプチドの合成を試みる場合において、従来の手法を用いる場合、次のような問題点があった。即ち、従来の化学合成法では、その材料として保護アミノ酸を用いる必要があり、一旦、化学合成し、脱保護精製して得られたペプチドは、もはや化学合成の材料として用いることはできなかった。このため、機能単位のペプチドを組み合わせて、さらに高度の機能のペプチドを合成しようとする場合は、もはや保護基をもたないペプチドは化学合成の材料として利用できないという欠点があった。精製して得られたペプチドを原料として、従来の化学合成で行われているような保護を行うことなく、ペプチド同士を結合できれば、ペプチド工学の分野において画期的な技術となる。このため、ペプチドの末端どうしを結合させる技術の開発が試みられるようになった。例えば、Tamら(C.F.Liu, C.Rao, J.P.Tam, J.American Chemical Society, 118, 307(1996))は、一般に化学反応の進みにくいペプチド間の反応を、まずアルデヒドとシステイン間にチオエステル結合中間体を形成させ(キャプチャー反応(Capture反応))、その後近接効果に基づいたアリルリアレンジメント反応(Aryl rearrangement反応)によりアミド結合生成に導いている。しかしながら、この方法により生成される結合部位は天然のペプチド結合と異なり、5員環の環状構造を持つことに難点がある。また同様な方法がいくつか開発されているが、いずれも結合により生じた構造はいわゆるペプチド結合、「-CO-NH-」、とは全く異なった構造となっている。さらに、その反応も複雑である(三原久和、化学、51巻6号、396ページ)。このように、現在まで、ペプチド同士を組み合わせて、新規なペプチドを合成する効果的な方法は皆無に等しい。
【0004】
【発明が解決しようとする課題】本発明は、ペプチド同士を直接結合し、新規なペプチドをブロック単位で合成することを可能にする方法を提供することを課題とする。
【0005】
【課題を解決するための手段】本発明者等は、タンパク質中のシステイン残基のスルフヒドリル基を化学修飾することによりシアノシステイン残基に転換し、シアノシステイン残基を介したペプチド鎖切断の反応のメカニズムを検討し、図1に示される反応が関与することを明らかにした。
【0006】図1中、反応1は、ヤコブソン(Jacobson)ら(G.R.Jacobson, M.H.Schaffer, G.R.Stark, T.C.Vanaman, J.Biological Chemistry, 248, 6583-6591(1973)に記載)により報告された反応で、水酸基がシアノシステイン残基のN末端側に隣接するアミノ酸に由来するカルボニル炭素を求核的に攻撃することにより起こるペプチド鎖の切断反応であり、反応2は、水酸基が酸・塩基触媒として働くことにより、チオシアノ基が脱離するβ脱離反応で、シアノシステイン残基がデヒドロアラニンに転換する反応である(Y.Degani, A.Patchornik, Biochemistry, 13, 1-11(1974)に記載)。反応3は、発明者らが、リジン−シアノシステインの配列を有するペプチドを開裂させた時に得られた切断生成物を詳細に解析した結果見出した反応である。この反応3においては、カルボキシ末端がラクタムを含む7員環構造となることから、反応3はリジンのεアミノ基がカルボニル炭素を求核的に攻撃した結果起こったものと考えられる。本発明者等は、分子間でも、一級アミンがシアノシステイン残基のN末端側に隣接するアミノ酸に由来するカルボニル炭素を求核的に攻撃することができるはずであると考えた(図4)。即ち、本発明者等は、反応4によってどのような一級アミン誘導体でも、シアノシステイン残基の前までの配列に結合させることができ、且つ、生じる結合は、「-CO-NH-」、即ちペプチド結合であるという着想を得た。そして、本発明者等は、図1の反応4が実際起こることを証明し、この機構に基づき2つのペプチド鎖を容易に結合させることができることを示し、本発明を完成した。具体的には、図2の反応4に示されるように、シアノシステイン残基を有するペプチド鎖「R1-CO-NH-C(CH2-SCN)-CO-NH-R2」にアミノ末端がフリーな別のペプチド鎖「NH2-R3」を作用させ、ペプチド鎖内のアミノ基にペプチド結合上のカルボニル炭素を求核攻撃させることにより、ペプチド間の結合反応を生じさせ、「R1-CO-NH-R3」の配列を持つペプチド鎖を合成できること示し、本発明を完成した。
【0007】すなわち、本発明は、一般式(1) R1-CO-NH-C(CH2-SCN)-CO-NH-R2で示されるシアノシステイン残基を有するペプチド性化合物に、一般式(2) NH2-R3で示される一級アミンを有する化合物を作用させることにより、一般式(3) R1-CO-NH-R3で示される化合物を生成させる方法〔式中、R1、R2、R3は、任意のアミノ酸配列を表す〕に関する。
【0008】本発明に係る反応の本質からして、ここで、R1、R2、及びR3の化学的形態は限定されないことは明白である。従って、本発明は、R1、R2、及びR3としては、非天然アミノ鎖を含むペプチド鎖及びその誘導体、アルキル鎖及びその誘導体、糖鎖及びその誘導体、脂肪酸及びその誘導体、ポリヌクレオチド及びその誘導体、ポリデオキシヌクレオチド及びその誘導体、ビタミン類及びその誘導体、各種抗生物質、などが含まれる他幅広い化学基が利用可能である。
【0009】
【発明の実施の形態】本発明によって、図2の反応式4のごとく、前記一般式(1)「R1-CO-NH-C(CH2-SCN)-CO-NH-R2」で示される化合物に、前記一般式(2)「NH2-R3」で示される化合物を反応させることにより、前記一般式(3)「R1-CO-NH-R3」で示される化合物を生成させることができる。ここで、一般式(1)で示される化合物は、一般式(4) R1-CO-NH-C(CH2-SH)-CO-NH-R2、即ち、R1とR2の間にシステイン残基を有する化合物のシステインのスルフヒドリル基を選択的にシアノ化することにより生成することができる。
【0010】シアノ化試薬としては、通常、2-ニトロ-5-チオシアノ-安息香酸(2-nitro-5-thiocyanobenzoic acid (NTCB))(Y.Degani, A.Patchornik, Biochemistry, 13, 1-11(1974)に記載)を用いる方法が簡便である。シアノ化の手法によって、本発明が制限を受けないことは明白である。NTCBは市販のものをそのまま用いることができる。NTCBを用いたシステインのスルフヒドリル基のシアノ化は、pH7〜9の間で効率よく行うことができ、且つ、遊離するが、チオニトロ安息香酸(thionitrobenzoate)の412nmの吸光度の増加(分子吸光係数=13,600)でシアノ化の反応効率を調べることができる。また、システインのスルフヒドリル基のシアノ化は、文献(J.Wood, & N. Catsimpoolas, J. biological Chemistry, 233, 2887(1963))記載の方法に従い, スルフヒドリル基のシアンによる直接的酸化で行うこともできる。従って、シアノ化の手法によって、本発明が制限を受けないことは明白である。
【0011】一般式(1)の化合物「R1-CO-NH-C(CH2-SCN)-CO-NH-R2」への、一般式(2)の化合物「NH2-R3」の反応は、弱アルカリ条件下(pH8〜10)に、室温で行うことができる。実施例1においては、「NH2-R3」としてグリシンアミドを用いた例を示す。この場合、図1R>1に示される反応1及び反応2が競争反応となるが、ペプチド結合生成反応が最も効率良く進むことが示されている。実施例2においては、pH10の条件を用いて、「NH2-R3」として、グリシン-L-アラニン-L-アラニン-L-チロシン-L-アラニンを用いた結果を示しているが、ペプチド結合反応の生成物が主反応生成物であることが示されている。この結果は、図1の反応4が実際に起こることを証明しており、さらに、この機構に基づき、2つのペプチド鎖を容易に結合させることができたことを実証している。しかしながら、本発明のペプチド鎖結合反応は、実施例で示した結合反応条件には、限定されない。即ち、図1の反応4は、反応の原料である一般式(1)の化合物「R1-CO-NH-C(CH2-SCN)-CO-NH-R2」及び一般式(2)の化合物「NH2-R3」が溶解し互いに溶液中で混じりあう条件でありさえすれば、進行する。原料を溶解する溶媒としては、水溶性の溶媒である、水、メタノール、エタノールなどのアルコール類の他ペプチドの溶解に用いられる、ジメチルホルムアミド(DMF)、ジメチルスルホキサイド(DMSO)などが可能であるが、本発明は、用いる溶媒によっては限定されないことは明白である。反応温度は、室温で高い反応効率が得られるが、用いる溶媒が、凍結もしくは沸騰しない温度範囲であれば問題なく用いることができる。
【0012】反応の追跡は、高速液体クロマトグラフィーを連結した質量分析装置を用いて、生成物を質量数で同定・帰属し、定量することにより行うことができる。実施例における生成物の同定・帰属・定量は、「LC10A型高速液体クロマトグラフィー」(島津製作所製)を連結した「PE Sciex API III質量分析装置」(パーキンエルマー社製)を用いて行ったが、生成物を正確に同定・帰属・定量できる方法であればどのような方法で行ってよく、本発明が反応の追跡方法に制限されることはないことは明らかである。
【0013】
【実施例】
[実施例1] N-アセチル-L-アラニン-L-アラニン-グリシン-(S-シアノ)L-システイン-L-アラニン(以下、「acY-A-A-G-cC-A」と略す。質量=622)とグリシンアミド(G-NH2)との結合反応ペプチド内のシアノシステインとそのN末端側に隣接するアミノ酸残基との間のペプチド結合内に存在するカルボニル炭素に対して、アミノ基による分子間求核攻撃が可能であると考えられた。そこで、ペプチドとして「acY-A-A-G-cC-A」を用い、アミノ基を有する分子としてグリシンアミドを用いて、反応を行った。具体的には、0.05Mリン酸と0.1Mホウ酸からなる緩衝液(pHは、7〜10まで変動させた)に、終濃度1mg/mlとなるように 「acY-A-A-G-cC-A」を加え、さらに終濃度10mg/mlとなるようにグリシンアミド(G-NH2)を加え、室温で3時間反応させた。なお、本実験の対照としてグリシンアミドの非存在下で反応を行った。本反応の場合、シアノシステインのN末端側に隣接するアミノ酸がグリシンであるため、図1の反応3は、起こり得ない。従って、本実施例において起こりうる反応経路は、図2で示される反応1、反応2、及び反応4である。また、それぞれの反応生成物である、「R1-COOH」,「R1-CO-NH-(C=CH2)-CO-NH-R2」, 及び「R1-CO-NH-R3」のそれぞれの質量数は、423、563、及び480である。
【0014】本実験におけるグリシンアミド存在下での結果を表1に、非存在下での結果を表2にそれぞれ示す。
【0015】
【表1】
──────────────────────────────────── 反応生成物の割合(グリシンアミドの存在下)
──────────────────────────────────── pH acY-A-A-G-cC-A 反応1の 反応2の 反応4の (未反応物) 生成物1 生成物2 生成物4(質量数 622 423 563 480 )──────────────────────────────────── 7 〜100(%) 〜0(%) 〜0(%) 〜0(%)
8 96.8 0 1.2 2.0 9 65.2 0 4.7 14.1 10 32.1 0.5 5.9 26.4────────────────────────────────────
【0016】
【表2】
──────────────────────────────────── 反応生成物の割合(グリシンアミドの非存在下)
──────────────────────────────────── pH acY-A-A-G-cC-A 反応1の 反応2の 反応4の (未反応物) 生成物1 生成物2 生成物4(質量数 622 423 563 480 )──────────────────────────────────── 7 〜100(%) 〜0(%) 〜0(%) 〜0(%)
8 89.6 9.2 1.2 0 9 65.2 32.0 2.8 0 10 32.1 53.0 14.9 0 ────────────────────────────────────表2から明らかなように、グリシンアミドの非存在下では、塩基性条件における水酸化物イオンが、ペプチド結合上のカルボニル炭素に対して求核攻撃を行う、反応1が主反応となって、ペプチドの切断が起こり、「acY-A-A-G」(質量数=423)と2-イミノチアゾリジン-4-カルボキシル-アラニン(2-iminothiazolidine-4-carboxylyl-alanine)が生成した。また、水酸化物イオンが塩基として作用しβ-脱離反応が起こる結果、ペプチドの切断が生じない反応2が副反応となり、N-アセチル-L-アラニン-L-アラニン-グリシン-L-デヒドロアラニン−L-アラニン(以下、「acY-A-A-G-dA-A」と略す。質量=563)が生成した。
【0017】一方、表1から明らかなように、グリシンアミド存在下では、グリシンアミド中のアミノ基が、ペプチド結合上のカルボニル炭素へ求核攻撃を行う反応4が主反応となって、この結果、ペプチドの切断と共に新たなペプチドの合成が引き起こされ、N-アセチル-L-アラニン-L-アラニン-グリシン-グリシンアミド(以下、「acY-A-A-G-G−NH2」と称する。質量数=480)が生成した。特に、高pH条件下では、主反応である反応4が促進されると共に、グリシンアミド非存在下の場合と比較して、副反応である反応2が阻害された。
【0018】[実施例2] N-アセチル-L-アラニン-L-アラニン-グリシン-(S-シアノ)L-システイン-L-アラニン(acY-A-A-G-cC-A)とグリシン-L-アラニン-L-アラニン-L-チロシン-L-アラニン(以下、「G-A-A-Y-A」と称する)とのペプチド結合反応実施例1において、グリシンアミド内のアミノ基による、ペプチド結合上のカルボニル炭素に対しての求核攻撃が可能であることが証明された。そこで、次に、グリシンアミドのような単純なアミノ酸誘導体に代えて、ペプチドである「G-A-A-Y-A」を用いて、同様の反応を行った。具体的には、0.05Mリン酸と0.1Mホウ酸からなる緩衝液に、終濃度1mg/mlとなるように 「acY-A-A-G-cC-A」を、終濃度10mg/mlとなるように「G-A-A-Y-A」を加え、室温で3時間反応させた。なお、緩衝液のpHは、実施例1で最もペプチド結合反応が促進されたpH10とした。この結果を表3に示す。
【0019】
【表3】
──────────────────────────────────── 反応生成物の割合──────────────────────────────────── acY-A-A-G-cC-A 反応1の 反応2の 反応4の (未反応物) 生成物1 生成物2 生成物4(質量数 622 423 563 857 )──────────────────────────────────── 60.1(%) 5.8(%) 12.4(%) 21.7(%) ────────────────────────────────────表3から明らかなように、グリシンアミドに代えて、ペプチドである「G-A-A-Y-A」を用いた場合でも、ペプチド内のグリシン残基のアミノ基による求核攻撃(反応4)により、新たなペプチド、N-アセチル-L-アラニン-L-アラニン-グリシン-グリシン-L-アラニン-L-アラニン-L-チロシン-L-アラニン(「acY-A-A-G-G-A-A-Y-A]、質量数=857)が生成した。
【0020】
【発明の効果】本発明により、保護基を用いずに、簡易かつ迅速にペプチドの合成を行う画期的な技術が提供された。これにより、機能単位のペプチドを組み合わせて、さらに高度の機能のペプチドを合成するなど、ペプチド合成の分野を中心に幅広い利用が期待される。
【図面の簡単な説明】
【図1】シアノシステイン残基を介したペプチド鎖切断に関与する反応の模式図である。
【図2】シアノシステイン残基を介したペプチド鎖切断に関与する反応のうち本発明において関与している反応の模式図である。
【特許請求の範囲】
【請求項1】 一般式R1-CO-NH-C(CH2-SCN)-CO-NH-R2で示されるシアノシステイン残基を有するペプチド性化合物に、一般式NH2-R3で示される一級アミンを有する化合物を作用させることにより、一般式R1-CO-NH-R3で示される化合物を生成させる方法。〔式中、R1、R2、R3は、任意のアミノ酸配列を表す〕
【請求項1】 一般式R1-CO-NH-C(CH2-SCN)-CO-NH-R2で示されるシアノシステイン残基を有するペプチド性化合物に、一般式NH2-R3で示される一級アミンを有する化合物を作用させることにより、一般式R1-CO-NH-R3で示される化合物を生成させる方法。〔式中、R1、R2、R3は、任意のアミノ酸配列を表す〕
【図1】


【図2】
【図2】
【公開番号】特開平10−45798
【公開日】平成10年(1998)2月17日
【国際特許分類】
【出願番号】特願平8−203771
【出願日】平成8年(1996)8月1日
【出願人】(000001144)工業技術院長 (75)
【上記1名の復代理人】
【弁理士】
【氏名又は名称】清水 初志 (外1名)
【出願人】(000000217)エーザイ株式会社 (102)
【上記1名の代理人】
【弁理士】
【氏名又は名称】清水 初志
【公開日】平成10年(1998)2月17日
【国際特許分類】
【出願日】平成8年(1996)8月1日
【出願人】(000001144)工業技術院長 (75)
【上記1名の復代理人】
【弁理士】
【氏名又は名称】清水 初志 (外1名)
【出願人】(000000217)エーザイ株式会社 (102)
【上記1名の代理人】
【弁理士】
【氏名又は名称】清水 初志
[ Back to top ]