
光電変換素子及びπ共役型有機ラジカル化合物

【課題】電解質層の腐食性が低く、かつ、光電変換効率に優れる光電変換素子を提供する。
【解決手段】半導体層121及び色素を含む半導体電極と、対電極140と、半導体電極と対電極との間に設けられた電解質層130とを含む光電変換素子110であって、電解質層が一般式(1)の化合物を含む。

(式(1)中、X1からX5で表される置換基は、それぞれ独立に電子吸引性基、水素、炭素数1〜6のアルキル基、アルコキシ基、フッ素置換アルコキシ基のいずれか)
【解決手段】半導体層121及び色素を含む半導体電極と、対電極140と、半導体電極と対電極との間に設けられた電解質層130とを含む光電変換素子110であって、電解質層が一般式(1)の化合物を含む。
(式(1)中、X1からX5で表される置換基は、それぞれ独立に電子吸引性基、水素、炭素数1〜6のアルキル基、アルコキシ基、フッ素置換アルコキシ基のいずれか)
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、光電変換素子及びπ共役型有機ラジカル化合物に関する。
【背景技術】
【0002】
無機化合物によって構成された材料、素子、デバイスを凌ぐ分子ナノエレクトロニクス材料の出現に伴い、有機化合物の機能がエレクトロニクス材料の主役として注目されている。有機化合物をエレクトロニクス材料として用いた有機デバイスの例としては、太陽電池や有機エレクトロルミネッセンス素子、電界効果トランジスタ(Field Effect Transistor:FET)等が挙げられる。これらは有機化合物の電気物性及び光物性を利用したデバイスであり、近年めざましい発展を遂げている。
【0003】
それらの発展に付随して、有機ラジカル化合物を利用した新しい技術開発が行われるようになってきた。有機ラジカル化合物は不対電子に由来する特異な電子・磁気特性を示し、特に強磁性が付与された化合物は、例えば、低重力の宇宙空間において使用する軽量磁石、無機の磁性フィラーと比べて比重の小さい磁性塗料、情報の高密度化が可能な記録材料、外部磁石によりコントロール可能なドラッグデリバリーシステム素材等への応用が期待できる。また、DNA塩基と類似の複素環構造を有し、さらにはラジカル酸化能を有しているので、DNA相互作用物質又はDNA切断剤等への応用も期待できる。
【0004】
実用的な有機ラジカルの代表例としては、ニトロキシドラジカルが挙げられる。ニトロキシドラジカルの電子構造は、不対電子が酸素原子上にある構造と窒素原子上にある構造の共鳴によって表される。さらに、ニトロキシドラジカルは酸素原子上の非共有電子対によりルイス塩基性を示すので、金属イオンとの配位能や水素結合のプロトン受容性を有する。このニトロキシドラジカルを利用した有機デバイスも近年盛んに報告されるようになり、特に、光電変換素子として様々な利用方法が提案されている。
【0005】
例えば、非特許文献1、特許文献1及び特許文献2には、腐食性の高い遊離のヨウ素化合物の代わりに2,2,6,6−テトラメチルピペリジン−N−オキシル(以下「TEMPO」ともいう。)又はその誘導体をレドックスメディエーターとして用いた色素増感太陽電池が記載されている。特許文献3には、配位子にラジカル部位を備えた光吸収材料が記載されている。特許文献4及び特許文献5には、ラジカル化合物からなる有機化合物層を電極と電荷輸送層の間に備えた光電変換素子が記載されている。
【0006】
ところで、TEMPOに代表される非共役型有機ラジカル化合物は、半占軌道(Singly Occupied Molecular Orbital:以下「SOMO」ともいう。)がニトロキシド部位に局在化している。そのために、酸化還元反応の可逆安定性には優れるものの、他の物質との相互作用という観点からは必ずしも最適な構造ではなく、光電変換素子の材料として更なる改良が求められている。特に、色素増感太陽電池のレドックスメディエーターとして用いる場合、錯体色素との相互作用において改善の必要性がある。
【0007】
一方、非特許文献2には、π共役型有機ラジカル化合物である2,2−ジフェニル−1,2−ジヒドロキノリン−1−オキシル(以下「DPQN」ともいう。)及びその誘導体が記載されている。しかしながら、従来公知の文献によるとπ共役平面による自己集積機能のみが研究対象となっていた。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2009−76369号公報
【特許文献2】特開2010−205704号公報
【特許文献3】特開2011−6665号公報
【特許文献4】特開2003−100360号公報
【特許文献5】特開2009−21212号公報
【非特許文献】
【0009】
【非特許文献1】Z. Zhang, P. Chen, T. N. Murakami, S. M. Zakeeruddin, M. Gratzel, Adv. Funct. Mater. 18, 341-346 (2008)
【非特許文献2】N. Yoshioka et al., Chem. Phys. Lett. 402, 11-16 (2005)
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は、上記事情に鑑みてなされたものであり、電解質層の腐食性が低く、かつ、光電変換効率に優れる光電変換素子及びそれを構成するπ共役型有機ラジカル化合物を提供することを目的とする。
【課題を解決するための手段】
【0011】
本発明者は、π共役型有機ラジカル化合物は、SOMOがπ共役平面に非局在化して存在しているため、他の物質との相互作用という観点から、光電変換素子として使用した場合には有利に働く可能性があるのではないかと考えた。そして、上記課題を解決するために鋭意研究を重ねた結果、特定のπ共役型有機ラジカル化合物が腐食性の低い新たな光電変換素子の材料として光電変換効率の向上に寄与することを見出し、本発明を完成するに至った。すなわち、本発明は下記のとおりである。
[1]半導体層及び色素を含む半導体電極と、対電極と、該半導体電極と該対電極との間に設けられた電解質層とを含む光電変換素子であって、該電解質層が、下記一般式(1)で示されるπ共役型有機ラジカル化合物を含む、上記光電変換素子。
【0012】
【化1】
(式(1)中、X1、X2、X3、X4、及びX5で表される置換基は、それぞれ独立に電子吸引性基、水素、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、又は炭素数1〜6のフッ素置換アルコキシ基のいずれかである。)
[2]前記一般式(1)中、X1、X2、X3、X4、及びX5で表される置換基のうち少なくとも1つは電子吸引性基であり、残余の置換基は、それぞれ独立に水素、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、又は炭素数1〜6のフッ素置換アルコキシ基のいずれかである[1]記載の光電変換素子。
[3]前記電子吸引性基が、F、Cl、NO2、CN、C6F5、及びCnF2n+1(nは1〜6の整数)からなる群からそれぞれ独立に選ばれた基である[2]記載の光電変換素子。
[4]前記色素が金属錯体色素である[1]〜[3]のいずれか一項記載の光電変換素子。
[5]下記一般式(1)で示されるπ共役型有機ラジカル化合物。
【0013】
【化2】
(式(1)中、X1、X2、X3、X4、及びX5で表される置換基のうち少なくとも1つは電子吸引性基であり、残余の置換基は、それぞれ独立に水素、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、又は炭素数1〜6のフッ素置換アルコキシ基のいずれかである。)
[6]前記電子吸引性基が、F、Cl、NO2、CN、C6F5、及びCnF2n+1(nは1〜6の整数)からなる群からそれぞれ独立して選ばれた基である[5]記載のπ共役型有機ラジカル化合物。
【発明の効果】
【0014】
本発明によると、電解質層の腐食性が低く、かつ、光電変換効率に優れる光電変換素子及びそれを構成するπ共役型有機ラジカル化合物を提供することができる。
【図面の簡単な説明】
【0015】
【図1】本発明の光電変換素子の一例の断面模式図。
【図2】実施例の光電変換素子のI−V曲線。
【図3】実施例の光電変換素子のI−V曲線。
【図4】参考例の光電変換素子のI−V曲線。
【図5】比較例の光電変換素子のI−V曲線。
【図6】実施例の光電変換素子のI−V曲線。
【図7】実施例の光電変換素子のI−V曲線。
【図8】実施例の光電変換素子のI−V曲線。
【図9】実施例の光電変換素子のI−V曲線。
【発明を実施するための形態】
【0016】
以下、本発明の実施態様(以下「本実施態様」ともいう。)について詳細に説明する。
(光電変換素子)
光電変換素子は、光エネルギーを電気エネルギーに変換する素子であり、色素増感太陽電池を包含する用語と定義する。
【0017】
本実施態様の光電変換素子は、半導体層及び色素を含む半導体電極と、対電極と、前記半導体電極と前記対電極との間に設けられた電解質層とを含むものであり、前記電解質層が下記一般式(1)で示されるπ共役型有機ラジカル化合物を含むことを特徴としている。
【0018】
【化3】
(式(1)中、X1、X2、X3、X4、及びX5で表される置換基は、それぞれ独立に電子吸引性基、水素、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、又は炭素数1〜6のフッ素置換アルコキシ基のいずれかである。)
ここで、前記一般式(1)で示されるπ共役型有機ラジカル化合物は、X1、X2、X3、X4、及びX5で表される置換基のうち少なくとも1つは電子吸引性基であり、残余の置換基は、それぞれ独立に水素、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、又は炭素数1〜6のフッ素置換アルコキシ基のいずれかである化合物が好ましい。また、前記電子吸引性基が、F、Cl、NO2、CN、C6F5、及びCnF2n+1(nは1〜6の整数)からなる群からそれぞれ独立に選ばれた基である化合物がより好ましい。
(π共役型有機ラジカル化合物)
一般式(1)で示されるπ共役型有機ラジカル化合物は、ジフェニル骨格の遮蔽効果によってNO部位が保護されるため、室温大気下においても化学的安定性が保たれている。また、ニトロキシドラジカルの窒素原子がπ共役平面の六員環構造上にあるため、酸化還元状態においてもπ共役平面が開環せず、長期安定性にも優れているという特徴がある。
【0019】
置換基として導入したX1、X2、X3、X4、及びX5は2つの役割を担っている。
【0020】
第一の役割は、π共役平面への不対電子密度の広がりを制御することである。π共役型有機ラジカル化合物は、元々SOMOがπ共役平面に拡張しているため、非共役型有機ラジカル化合物と比較して光電変換素子としての使用時における他の物質との相互作用には有利であると考えられる。そこで、X1、X2、X3、X4、又はX5に置換基として電子吸引性基を導入することによりπ共役平面の不対電子密度を広げることが、他の物質との相互作用を一段と高める効果があるので好ましい。なお、電子吸引性基の導入位置はX1、X2、又はX3の位置がより好ましく、X2の位置が特に好ましい。また、前記電子吸引性基が、F、Cl、NO2、CN、C6F5、及びCnF2n+1(nは1〜6の整数)からなる群からそれぞれ独立に選ばれた基であることがより好ましい。
【0021】
第二の役割は、溶媒への溶解性を制御することである。置換基を導入すると分子容が大きくなるため、π共役平面の自己集積機能を低減させる効果がある。言い換えると、結晶性や溶解性等の物理化学的性質を制御することが可能になる。なお、第二の役割の場合には、電子吸引性基に限らず効果が認められるが、導入位置は第一の役割への影響が少ないX4、又はX5の位置が好ましい。
(π共役型有機ラジカル化合物の製造方法)
本実施態様におけるπ共役型有機ラジカル化合物は、下記2通りの反応ルートのいずれか一方により製造することができる。なお、反応式中、X1、X2、X3、X4、及びX5は前記のとおりである。
[反応ルート1]
【0022】
【化4】
[反応ルート2]
【0023】
【化5】
まず、上記一般式(2)で示される反応ルート1の製造方法について説明する。
【0024】
(i)の反応では、氷冷したm−クロロ過安息香酸のクロロホルム溶液を、同じく氷冷したキノリン誘導体(2A)のクロロホルム溶液に滴下し、その後、m−クロロ過安息香酸をアルカリ水溶液によって加水分解する。アルカリ水溶液として特に制限はないが、水酸化ナトリウム水溶液であることが好ましい。分液による水洗を経てクロロホルム相を回収し、脱水剤を加えて乾燥させる。脱水剤として特に制限はないが、無水硫酸ナトリウム又は無水硫酸マグネシウムであることが好ましい。溶媒を除去後、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製して化合物(2B)を得る。シリカゲルカラムクロマトグラフィーの展開溶媒として特に制限はないが、例えば、酢酸エチル:n−ヘキサン=1:1(体積比)とした展開溶媒等が挙げられる。
【0025】
(ii)の反応では、化合物(2B)を脱水テトラヒドロフランに溶解させ、不活性ガス置換する。不活性ガスとして特に制限はないが、アルゴン又は窒素であることが好ましい。これに置換基X4を有する4−置換フェニルマグネシウムブロミドを含むテトラヒドロフラン溶液を滴下して攪拌する。なお、反応温度について特に制限はないが、0〜60℃であることが好ましく、5〜50℃であることがより好ましく、10〜40℃であることがさらに好ましい。この反応は、低温では反応速度が低下し、高温では脱水反応が併発して酸素が取れた2−フェニルキノリン誘導体になるため、反応温度を前記の範囲内に保つことによって反応収率を高めることができる。反応後は、中和剤によって中和する。中和剤として特に制限はないが、飽和塩化アンモニウム水溶液であることが好ましい。分液による水洗を経てクロロホルム相を回収し、脱水剤を加えて乾燥させる。脱水剤として特に制限はないが、無水硫酸ナトリウム又は無水硫酸マグネシウムであることが好ましい。溶媒を除去後トルエンに溶解して沸点還流し、トルエンを除去した後に得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製して化合物(2C)を得る。シリカゲルカラムクロマトグラフィーの展開溶媒として特に制限はないが、例えば、酢酸エチル:n−ヘキサン=1:1(体積比)とした展開溶媒等が挙げられる。
【0026】
(iii)の反応では、化合物(2C)を脱水テトラヒドロフランに溶解させ、不活性ガス置換する。不活性ガスとして特に制限はないが、アルゴン又は窒素であることが好ましい。これに室温で置換基X5を有する4−置換フェニルマグネシウムブロミドを含むテトラヒドロフラン溶液を滴下して攪拌する。なお、反応温度について特に制限はないが、0〜60℃であることが好ましく、5〜50℃であることがより好ましく、10〜40℃であることがさらに好ましい。この反応は、低温では反応速度が低下し、高温では脱水反応が併発して酸素が取れた2−フェニルキノリン誘導体になるため、反応温度を前記の範囲内に保つことによって反応収率を高めることができる。反応後は、中和剤によって中和する。中和剤として特に制限はないが、飽和塩化アンモニウム水溶液であることが好ましい。なお、この状態ではN−OH体(2D)となっていると推定されるが、不安定であるためキャラクタリゼーションは困難である。したがって、(iii)の反応に続けて(iv)の反応を行うことが好ましい。
【0027】
(iv)の反応では、(iii)で合成した化合物(2D)をクロロホルムに溶解して暗所に静置することにより空気酸化させる。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製することによってπ共役型有機ラジカル化合物(2E)を得ることができる。シリカゲルカラムクロマトグラフィーの展開溶媒として特に制限はないが、例えば、クロロホルム等が挙げられる。
【0028】
次に、上記一般式(3)で示される反応ルート2の製造方法について説明する。
【0029】
(v)の反応では、置換基X1及びX2を有するアニリン誘導体(3A)にエタノールを加え、加熱溶解させる。これに置換基X4を有するベンズアルデヒド誘導体を添加して沸点還流する。析出した粗結晶を濾別し、n−ヘキサン洗浄によって精製して化合物(3B)を得る。
【0030】
(vi)の反応では、化合物(3B)にベンゼンを加え溶解させる。蒸留したアルキルビニルエーテルと三フッ化ホウ素ジエチルエーテル錯体を加えて加熱攪拌する。アルキルビニルエーテルとして特に制限はないが、エチルビニルエーテルであることが好ましい。反応温度について特に制限はないが、25〜60℃であることが好ましく、30〜50℃であることがより好ましく、35〜45℃であることがさらに好ましい。攪拌時間は1〜72時間であることが好ましく、5〜48時間であることがより好ましく、10〜24時間であることがさらに好ましい。なお、(vi)の反応は、溶媒にアセトニトリルを用い、硝酸二アンモニウムセリウムを加えて攪拌しても進行する。室温に戻してアルカリ水溶液で中和し、クロロホルム溶液を加え、分液の後、有機相を回収する。アルカリ水溶液として特に制限はないが、水酸化ナトリウム水溶液であることが好ましい。その後、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製して化合物(3C)を得る。シリカゲルカラムクロマトグラフィーの展開溶媒として特に制限はないが、例えば、クロロホルム等が挙げられる。
【0031】
(vii)の反応では、化合物(3C)にトルエンを加え溶解させる。ジエチルアゾジカルボキシレートを加え沸点還流する。アルカリ水溶液で中和し、分液の後、有機相を回収する。アルカリ水溶液として特に制限はないが、水酸化ナトリウム水溶液であることが好ましい。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製して化合物(3D)を得る。シリカゲルカラムクロマトグラフィーの展開溶媒として特に制限はないが、例えば、クロロホルム:n−ヘキサン=1:1(体積比)とした展開溶媒等が挙げられる。なお、X2がI等のハロゲンである場合には、炭素数1〜6のアルコキシ基又は炭素数1〜6のフッ素置換アルコキシ基への置換が可能である。具体的には、例えば、X2がI等のハロゲンである化合物(3D)に、炭素数1〜6のアルコール又は炭素数1〜6のフッ素置換アルコール、ヨウ化銅、炭酸セシウム、及び2−オキソシクロヘキサカルボン酸エチルを加えて沸点還流する。反応時間は1〜72時間であることが好ましく、5〜48時間であることがより好ましく、10〜24時間であることがさらに好ましい。その後、分液による水洗を経て有機相を回収し、脱水剤を加えて乾燥させる。脱水剤として特に制限はないが、無水硫酸ナトリウム又は無水硫酸マグネシウムであることが好ましい。溶媒を除去後に得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製してX2の置換体が得られる。シリカゲルカラムクロマトグラフィーの展開溶媒として特に制限はないが、例えば、クロロホルム:n−ヘキサン=5:1(体積比)とした展開溶媒等が挙げられる。
【0032】
(viii)の反応では、氷冷したm−クロロ過安息香酸のクロロホルム溶液に、同じく氷冷した化合物(3D)のクロロホルム溶液に加え、その後、m−クロロ過安息香酸をアルカリ水溶液によって加水分解する。アルカリ水溶液として特に制限はないが、水酸化ナトリウム水溶液であることが好ましい。分液による水洗を経てクロロホルム相を回収し、脱水剤を加えて乾燥させる。脱水剤として特に制限はないが、無水硫酸ナトリウム又は無水硫酸マグネシウムであることが好ましい。溶媒を除去後、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製して化合物(3E)を得る。シリカゲルカラムクロマトグラフィーの展開溶媒として特に制限はないが、例えば、酢酸エチル:n−ヘキサン=1:1(体積比)とした展開溶媒等が挙げられる。
【0033】
(ix)の反応では、化合物(3E)を脱水テトラヒドロフランに溶解させ、不活性ガス置換する。不活性ガスとして特に制限はないが、アルゴン又は窒素であることが好ましい。これに室温で置換基X5を有する4−置換フェニルマグネシウムブロミドを含むテトラヒドロフラン溶液を滴下して攪拌する。なお、反応温度について特に制限はないが、0〜60℃であることが好ましく、5〜50℃であることがより好ましく、10〜40℃であることがさらに好ましい。この反応は、低温では反応速度が低下し、高温では脱水反応が併発して酸素が取れた2−フェニルキノリン誘導体になるため、反応温度を前記の範囲内に保つことによって反応収率を高めることができる。反応後は、中和剤によって中和する。中和剤として特に制限はないが、飽和塩化アンモニウム水溶液であることが好ましい。なお、この状態ではN−OH体(3F)となっていると推定されるが、不安定であるためキャラクタリゼーションは困難である。(ix)の反応に続けて(x)の反応に進むことが好ましい。
【0034】
(x)の反応では、化合物(3F)をクロロホルムに溶解して暗所に静置することにより空気酸化させる。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製することによってπ共役型有機ラジカル化合物(3G)を得ることができる。シリカゲルカラムクロマトグラフィーの展開溶媒として特に制限はないが、例えば、クロロホルム等が挙げられる。
【0035】
本発明におけるπ共役型有機ラジカル化合物の具体例を以下に例示する。
【0036】
【化6】
【0037】
【化7】
【0038】
【化8】
【0039】
【化9】
【0040】
【化10】
【0041】
【化11】
【0042】
【化12】
【0043】
【化13】
【0044】
【化14】
【0045】
(電解質層)
本実施態様(図1)における電解質層130は、前記のπ共役型有機ラジカル化合物と液体状又は固体状の電解質を含む。電解質層100重量部に対し、前記のπ共役型有機ラジカル化合物が1〜50重量部含まれることが好ましく、1.5〜40重量部含まれることがより好ましく、2〜30重量部含まれることが特に好ましい。この範囲にあることによって、光電変換素子の電解質層として必要な電荷輸送機能が確保され、かつ、析出や濃度ムラ等の影響なく光電変換効率の向上を達成することができる。
【0046】
本実施態様における液体状電解質として特に制限はないが、溶媒及び添加剤から構成されることが好ましい。
【0047】
前記溶媒としては、例えば、アセトニトリル、メトキシアセトニトリル、3−メトキシプロピオニトリル、ベンゾニトリル等のニトリル系溶媒;N−メチルピロリドン、N,N−ジメチルホルムアミド、3−メチル−2−オキサゾリジノン等の含窒素溶媒;γ−ブチロラクトン、バレロラクトン等のラクトン系溶媒;エチレンカーボネート、ジエチルカーボネート、ジメチルカーボネート、エチルメチルカーボネート、プロピレンカーボネート等のカーボネート系溶媒;テトラヒドロフラン、ジオキサン、ジエチルエーテル、エチレングリコールジアルキルエーテル等のエーテル系溶媒;メタノール、エタノール、イソプロピルアルコール、ポリプロピレングリコールモノアルキルエーテル等のアルコール系溶媒;ジメチルスルホキシド、スルホラン等の非プロトン性極性溶媒;等の溶媒が挙げられ、これらはそれぞれ単独で、又は2種以上混合して使用することができる。
【0048】
前記溶媒として、イオン液体、即ち溶融塩を用いることもできる。さらに、液体状電解質が常温溶融塩を含んでもよい。イオン液体としては、「Inorg. Chem.」1996, 35, p1168-1178、「Electrochemistry」2002.2, p130-136、特表平9−507334号公報、特開平8−259543号公報等に開示されている公知の電池や太陽電池等において一般的に使用することができるものであれば、特に限定されない。このうち、室温(25℃)より低い融点を有する塩か、又は室温よりも高い融点を有しても、他の溶融塩や溶融塩以外の添加物を溶解させることにより室温で液状化する塩が好ましく用いられる。
【0049】
具体的には、溶融塩のカチオンとしては、アンモニウム、イミダゾリウム、オキサゾリウム、チアゾリウム、ピペリジニウム、ピラゾリウム、イソオキサゾリウム、チアジアゾリウム、オキサジアゾリウム、トリアゾリウム、ピロリジニウム、ピリジニウム、ピリミジニウム、ピリダジニウム、ピラジニウム、トリアジニウム、ホスホニウム、スルホニウム、カルバゾリウム、インドリウム、及びこれらの誘導体が好ましく、アンモニウム、イミダゾリウム、ピリジニウム、ピペリジニウム、ピラゾリウム、スルホニウムが特に好ましい。
【0050】
また、溶融塩のアニオンとしては、AlCl4−、Al2Cl7−等の金属塩化物、PF6−、BF4−、CF3SO3−、N(CF3SO2)2−、CF3COO−等のフッ素含有物、NO3−、CH3COO−、C6H11COO−、CH3OSO3−、CH3OSO2−、CH3SO3−、CH3SO2−、(CH3O)2PO2−等の非フッ素化合物、塩素、臭素、ヨウ素等のハロゲン化物等が挙げられる。
【0051】
溶融塩は、各種文献や公報で公知の方法により合成することができる。4級アンモニウム塩を例に挙げると、第一段階として3級アミンにアルキル化剤としてアルキルハライドを用いてアミンの4級化を行い、第二段階としてハライドアニオンから目的のアニオンへイオン交換を行うという方法を用いることができる。あるいは、3級アミンを目的のアニオンを有する酸と反応させて一段階で目的の化合物を得る方法がある。
【0052】
溶媒としては、イオン伝導度の高いニトリル化合物又は常温溶融塩を用いることがより好ましい。
【0053】
本発明におけるπ共役型有機ラジカル化合物は、光電変換過程における酸化反応によってカチオン状態となる。このカチオン状態を安定化させるために、添加剤として塩を添加することも可能である。
【0054】
前記添加剤として用いる塩は、例えば、カチオンとして、リチウム、ナトリウム、カリウム、セシウム、カルシウム、ニトロソニウム、アンモニウム、テトラアルキルアンモニウム、イミダゾリウム、オキサゾリウム、チアゾリウム、ピペリジニウム、ピラゾリウム、イソオキサゾリウム、チアジアゾリウム、オキサジアゾリウム、トリアゾリウム、ピロリジニウム、ピリジニウム、ピリミジニウム、ピリダジニウム、ピラジニウム、トリアジニウム、ホスホニウム、スルホニウム、カルバゾリウム、インドリウム、及びこれらの誘導体が挙げられ、リチウム、ニトロソニウム、アンモニウム、イミダゾリウム、ピリジニウム、ピペリジニウム、ピラゾリウム、スルホニウムが好ましい。
【0055】
また、アニオンとしては、PF6−、BF4−、CF3SO3−、N(CF3SO2)2−、F(HF)j−(jは整数)、CF3COO−等のフッ素含有物、NO3−、CH3COO−、C6H11COO−、CH3OSO3−、CH3OSO2−、CH3SO3−、CH3SO2−、(CH3O)2PO2−、SbCl6−等の非フッ素化合物、ヨウ素、臭素等のハロゲン化物等が挙げられる。その他、具体的な組合せとして、フェロシアン酸塩−フェリシアン酸塩、フェロセン−フェリシニウムイオン等の金属錯体、ポリ硫化ナトリウム、アルキルチオール−アルキルジスルフィド等の硫黄化合物、ビオロゲン色素、ヒドロキノン−キノン等も挙げられる。これらの塩は、単独の組み合わせであっても混合であってもよい。なお、前記電解質層における前記塩濃度は、0.05〜20mol/lであることが好ましく、0.1〜15mol/lであることがさらに好ましい。この範囲にあることによって、光電変換素子の電解質層として必要な電荷輸送機能が確保され、かつ、析出や濃度ムラ等の影響なく光電変換効率の向上を達成することができる。
【0056】
本発明における固体状電解質として特に制限はないが、例えば、ポリマー添加、オイルゲル化剤添加、微粒子添加、多官能モノマー類を含む重合、ポリマー架橋反応等の手法により液体状電解質をゲル化又は固体化した電解質が挙げられる。ポリマー添加としては、例えば、ポリアクリロニトリル、ポリフッ化ビニリデン等の添加が挙げられる。オイルゲル化剤添加としては、例えば、ジベンジルデン−D−ソルビトール、コレステロール誘導体、アミノ酸誘導体、トランス−(1R,2R)−1,2−シクロヘキサンジアミンのアルキルアミド誘導体、アルキル尿素誘導体、N−オクチル−D−グルコンアミドベンゾエート、双頭型アミノ酸誘導体、4級アンモニウム誘導体等の添加が挙げられる。
【0057】
本発明における電解質層の形成方法として特に制限はなく、例えば、マイクログラビアコーティング、ディップコーティング、スクリーンコーティング、スピンコーティング等を用いることができる。なお、固体状電解質の場合は、例えば、任意の溶媒によって溶液とした後、上記方法を用いて電極に塗工し、任意の温度に加熱して溶媒を蒸発させればよい。
【0058】
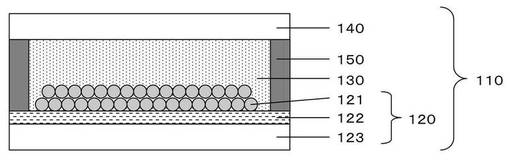
(半導体電極)
本実施態様(図1)における半導体電極120は、導電性基板とその上に形成された半導体層121からなり、導電性基板及び半導体層が光電変換素子110の外側から内側に向かってこの順に積層している。また、半導体層121には光増感剤として機能する色素が含まれている。
【0059】
前記導電性基板は、電極基材123と透明導電層122からなり、透明導電層の面が半導体層に接する。電極基材に用いられる材料は、透明であれば特に限定されるものではない。具体的には、ガラスや強化ガラス等のガラス類、ポリカーボネート、ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリエチレンナフタレート、ポリエチレンスルフィド、ポリスチレン、ポリメチルメタクリレート、ポリメタクリレート、アクリル樹脂、ポリ塩化ビニル等、トリアセチルセルロース、ポリイミド、環状ポリオレフィン、ポリエチレン、ポリプロピレン等のポリオレフィン系等のプラスチックフィルムを用いることができる。
【0060】
前記電極基材の厚さは、材料がプラスチックフィルムの場合は、10〜500μmが好ましく、50〜200μmがより好ましく、100〜200μmが特に好ましい。ガラス類の場合は、0.1〜5mmが好ましく、0.5〜4mmがより好ましく、0.5〜2.5mmが特に好ましい。また、電極基材は光の入射面として用いる場合、可視光域の透過率が85%以上であって、耐候性に優れ、かつ指示材として耐えうる強度を持つことが好ましい。このような電極基材は、必要に応じて表面がコロナ処理、プラズマ処理、薬品処理等によって改質されたものであってもよい。
【0061】
前記透明導電層としては、公知の可視光領域の吸収が少なく導電性のある透明導電材料を用いることができる。透明導電材料としては、錫をドープした酸化インジウム(ITO)、亜鉛をドープした酸化インジウム(IZO)、フッ素やインジウムやアンチモン等をドープした酸化錫、アルミニウムやガリウム等をドープした酸化亜鉛等が好ましい。また、銀又は銀合金(AgAuCu等)をITO、TiO2、ZnO等で挟んだ3層型透明導電材料も挙げられる。これらの中でも、特にITO又はフッ素をドープした酸化錫を使用することが好ましい。
【0062】
前記透明導電層の形成方法としては、真空蒸着法、反応性蒸着法、イオンビームアシスト蒸着法、スパッタリング法、イオンプレーティング法、プラズマCVD法等の真空製膜プロセスによることができる。しかしこれらの例に限定されることはなく、いかなる成膜方法であっても構わない。透明導電層の厚さは50nm〜1μmが好ましい。条件にもよるが、ITOの場合は、100〜400nmの膜厚が好ましく、フッ素ドープ酸化錫の場合は、300〜900nmが好ましい。透明導電層は、可視光域の透過率が65%以上であることが好ましい。
【0063】
本実施態様における半導体層を構成する材料としては、n型又はp型半導体の性質を示す金属酸化物等が挙げられ、例えば、亜鉛、ニオブ、錫、チタン、バナジウム、インジウム、タングステン、タンタル、ジルコニウム、モリブデン、マンガン、鉄、銅、ニッケル、イリジウム、ロジウム、クロム、ルテニウムの酸化物等が挙げられる。また、SrTiO3、CaTiO3、BaTiO3、MgTiO3、SrNb2O6のようなペロブスカイト、硫化カドミウム等、又は、これらの複合酸化物若しくは酸化物混合物等も使用することができる。これらの半導体材料は単独で用いることも2種類以上を混合して用いることもできる。
【0064】
これらの中でも、変換効率、安定性、安全性の観点からは、半導体層が、酸化チタンを含む半導体材料により構成されていることが好ましい。酸化チタンとして、さらに具体的には、アナターゼ型酸化チタン、ルチル型酸化チタン、無定形酸化チタン、メタチタン酸、オルソチタン酸等の種々の酸化チタン、含酸化チタン複合体等が挙げられる。その中でも、光電変換の安定性をさらに向上させる観点からは、アナターゼ型酸化チタンであることが好ましい。また、半導体層が、多孔性のチタニアであってもよい。
【0065】
半導体層の形状としては、半導体微粒子等を焼結することにより得られる多孔性半導体層、ゾル−ゲル法・スパッタ法・スプレー熱分解法等により得られる薄膜状半導体層等が挙げられる。また、その他繊維状半導体層や針状晶からなる半導体層としてもよい。半導体層の形状は、光電変換素子の使用目的に応じて、適宜選択することができる。
【0066】
このうち、色素吸着量等の観点からは、多孔性半導体層、針状晶からなる半導体層等比表面積の大きな半導体層が好ましい。さらに、半導体微粒子の粒径により入射光の利用率等を調整できるという観点からは、半導体層として半導体微粒子から形成される多孔性半導体層を用いることが好ましい。
【0067】
また、半導体層は、単層であっても多層であってもよい。多層にすることによって、充分な厚さの半導体層をさらに容易に形成することができる。
【0068】
また、半導体微粒子から形成される多孔性の多層半導体層は、半導体微粒子の平均粒径の異なる複数の半導体層からなってもよい。たとえば、光入射側に近い方の半導体層(第1半導体層)の半導体微粒子の平均粒径を、光入射側から遠い方の半導体層(第2半導体層)より小さくすることにより、第1半導体層で多くの光を吸収させるとともに、第1半導体層を通過した光を第2半導体層で効率よく散乱させて第1半導体層に戻し、第1半導体層で吸収させることにより、全体の光吸収率をより一層向上させることができる。
【0069】
半導体層の膜厚は、特に限定されるものではないが、透過性、変換効率等の観点より、0.5〜45μmが好ましい。半導体層の比表面積は、多量の色素を吸着によって含ませる観点から、10〜200m2/gが好ましい。
【0070】
また、多孔性の半導体層に色素を吸着させた構成として、電解質中のイオンがさらに充分に拡散して電荷輸送が行われるためには、多孔性の半導体層空隙率を40〜80%とすることが好ましい。なお、空隙率とは、半導体層の体積のうち、半導体層中の空隙が占める体積の割合を%で示したものである。
【0071】
次に、前記半導体層の形成方法について、多孔性半導体層を例にとって説明する。多孔性半導体層は、例えば、半導体微粒子を高分子等の有機化合物及び分散剤とともに、有機溶媒や水等の分散媒に加えて懸濁液を調製し、この懸濁液を透明導電層上に塗布し、これを乾燥、焼成することによって形成する。
【0072】
半導体微粒子とともに分散媒に有機化合物を添加しておくと、焼成時に有機化合物が燃焼して多孔性半導体層内にさらに充分な隙間を確保することが可能となる。また焼成時に燃焼する有機化合物の分子量や添加量を制御することで空隙率を変化させることができる。なお、有機化合物の種類や量は、使用する微粒子の状態、懸濁液全体の総重量等により適宜選択し調整することができる。ただし、半導体微粒子の割合が懸濁液全体の総重量に対して10wt%以上のときは、作製した膜の強度をより一層充分に強くすることができ、半導体微粒子の割合が懸濁液全体の総重量に対して40wt%以下であれば、空隙率が大きな多孔性半導体層をより一層安定的に得ることができるため、半導体微粒子の割合は懸濁液全体の総重量に対して10〜40wt%であることが好ましい。
【0073】
前記の半導体微粒子としては、適当な平均粒径、たとえば、1〜500nm程度の平均粒径を有する単一半導体又は化合物半導体の粒子等が用いられる。その中でも比表面積を大きくするという点からは、1〜50nm程度の平均粒径のものが望ましい。また入射光の利用率を高めるために、200〜400nm程度の平均粒径の比較的大きな半導体粒子を添加してもよい。
【0074】
また、半導体微粒子の製造方法としては、水熱合成法等のゾル−ゲル法、硫酸法、塩素法等が挙げられ、目的の微粒子を製造できる方法であればどんな方法を用いてもよいが、結晶性の観点からは、水熱合成法により合成することが好ましい。
【0075】
前記の有機化合物は、懸濁液中に溶解し、焼成するときに燃焼して除去できるものであれば何でも用いることができる。たとえば、ポリエチレングリコール、エチルセルロース等の高分子が挙げられる。懸濁液の分散媒としては、エチレングリコールモノメチルエーテル等のグライム系溶媒;イソプロピルアルコール等のアルコール類;及びイソプロピルアルコール/トルエン等の混合溶媒;並びに水等が挙げられる。
【0076】
懸濁液の塗布方法としては、ドクターブレード法、スキージ法、スピンコート法、スクリーン印刷法等公知の方法が挙げられる。その後、塗膜の乾燥、焼成を行う。乾燥と焼成の条件は、たとえば大気下又は不活性ガス雰囲気下、50〜800℃程度の範囲内で、10秒〜12時間程度とする。この乾燥及び焼成は、単一の温度で1回又は温度を変化させて2回以上行うことができる。
【0077】
なお、ここでは、多孔性半導体層の形成方法について詳述したが、他の種類の半導体層も種々の公知の方法を用いて形成することができる。例えば、金属酸化物の成膜には、形成したい金属酸化物に対応する金属、金属酸化物、金属亜酸化物等を蒸着源として電子ビームやプラズマ銃による加熱を用いた蒸着法、あるいは酸素ガスを導入しながら蒸着を行なう反応性蒸着法を用いることができる。成膜圧力は用いる蒸着源の種類によって異なるが、1×10−2〜1Paの範囲で行うことが好ましい。成膜の際、任意のガスを用いたプラズマやイオン銃、ラジカル銃等でアシストを行ってもよい。基板温度は、−50℃〜600℃の間で任意に選択することができるが、多孔性を高く保つためには300℃以下であることが好ましい。また目的の金属酸化物によっては、スパッタリング法、イオンプレーティング、CVD等の真空成膜法を用いてもよい。また基材にプラスチックフィルムを用いた場合には、ロールトゥロール方式で成膜すればより高い生産性を得ることができる。
【0078】
以上で得られた金属酸化物膜は、プラズマ処理、コロナ処理、UV処理、薬品処理等の任意の方法で表面処理することができる。また、熱による焼成や圧縮機を用いた加圧処置、レーザアニーリング等の任意の手段を用いて後処理することもできる。
【0079】
(色素)
本実施態様において半導体層に含まれる色素は、光増感剤として機能する。具体的には、色素は種々の可視光領域及び赤外光領域に吸収を持つものであって、吸収係数が大きくかつ繰り返しの酸化還元に対して安定であることが好ましい。また、半導体層に強固に吸着させるために、色素分子中にカルボキシル(COOH)基、アルコキシ基、ヒドロキシル基、ヒドロキシアルキル基、スルホン酸基、エステル基、メルカプト基、ホスホニル基、アミド基、アミノ基、カルボニル基、リン酸基等のインターロック基を有するものが好ましい。この中でも、色素を多孔質半導体表面にさらに安定的に吸着させる観点からは、カルボキシル基を有するものが特に好ましい。
【0080】
インターロック基は、色素の励起状態のエネルギー帯と半導体の伝導帯との間の電子移動を容易にする電気的結合を供給するものである。これらインターロック基を含有する色素としては、たとえば、Ru金属錯体色素(ルテニウムビピリジン系金属錯体色素、ルテニウムターピリジン系金属錯体色素、ルテニウムクォーターピリジン系金属錯体色素等)、Os金属錯体色素、Fe金属錯体色素、Cu金属錯体色素、Pt金属錯体色素、Re金属錯体色素等の金属錯体色素、シアニン系色素、メロシアニン系色素、アゾ系色素、キノン系色素、キノンイミン系色素、キナクリドン系色素、スクアリリウム系色素、トリフェニルメタン系色素、キサンテン系色素、ポルフィリン系色素、フタロシアニン系色素、ペリレン系色素、インジゴ系色素、ナフタロシアニン系色素、クマリン系色素、マーキュロクロム系色素、シアニジン系色素、ローダミン系色素等が挙げられる。その中でも、光電変換反応の安定性の観点からは、金属錯体色素が好ましく、Ru金属錯体色素がより好ましい。
【0081】
前記の半導体層に前記の色素を吸着させる方法としては、たとえば導電性基板上に形成された半導体層を、色素を溶解した溶液に浸漬する方法が挙げられる。色素を溶解するために用いる溶媒は、例えば、エタノール等のアルコール類;アセトン等のケトン類;ジエチルエーテル、テトラヒドロフラン等のエーテル類;アセトニトリル等の窒素化合物;クロロホルム等のハロゲン化脂肪族炭化水素;ヘキサン等の脂肪族炭化水素、ベンゼン等の芳香族炭化水素;酢酸エチル等のエステル類等が挙げられる。またこれらの溶媒は2種類以上を混合して用いてもよい。
【0082】
溶液中の色素濃度、使用する色素及び溶媒の種類は適宜調整することができるが、吸着機能を向上させる観点からは、ある程度高濃度である方が好ましく、例えば、5×10−5mol/l以上の濃度であればよい。
【0083】
色素を溶解した溶液中に半導体層を形成した導電性基板を浸漬する際、溶液及び雰囲気の温度及び圧力は特に限定されるものではなく、例えば室温程度、かつ大気圧下が挙げられ、浸漬時間は使用する色素、溶媒の種類、溶液の濃度等により適宜調整することができる。なお、効果的に行うには加熱下にて浸漬を行えばよい。これにより、半導体層に色素をさらに効率よく吸着させることができる。また、色素を吸着する際、色素の吸着状態を制御するため、色素を溶解した溶液にデオキシコール酸やグアニジンチオシアネート、tert−ブチルピリジン、N−メチルベンズイミダゾール、N−(n−ブチル)ベンズイミダゾール、エタノール等の有機化合物を加えてもよい。
【0084】
(対電極)
本実施態様(図1)における対電極140としては、導電性被膜を形成できる程度の平滑性を備え、かつ封止材を挟み込む程度の強度を有するものであれば、特に限定されるものではなく、無機系材料、有機系材料、金属系材料等の材質を問わず用いることができる。具体的には、白金、金、銀等の貴金属材料、銅、アルミニウム、炭素等の導電性材料が挙げられる。腐食や長期耐久性を考慮すると、白金、金、銀等の貴金属材料や炭素が好ましく、さらにこれらの貴金属又は炭素を蒸着したガラス又はプラスチックフィルム等を使用することも経済性の観点から好ましい。また、可視光透過性を有する光電変換素子を得るために、ITO、酸化錫、フッ素ドープされた酸化錫等の透明導電膜を使用することもできる。なお、対電極の厚さは、材質にもよるが、強度と製品の軽量性を考慮して10〜3000μmが好ましく、50〜1000μmがより好ましい。
【0085】
本発明における対電極には、必要に応じて導電性触媒層を設けてもよい。導電性触媒層としては、任意の導電性材料を用いることができ、白金、金、銀、銅等の金属、又は炭素等を挙げることができる。また、ポリアニリン、ポリチオフェン、PEDOT、ポリピロール等の導電性材料を用いることもできる。これらを形成する際には、透明導電層と同様の真空成膜法、あるいはこれら材料の微粒子をペーストにしたものをウェットコーティングする方法を用いることができる。
【0086】
前記導電性触媒層の厚さは、0.1〜2000nmが好ましく、1〜500nmがより好ましく、5〜200nmがさらに好ましい。なお、腐食性の高い遊離のヨウ素化合物等が含まれる場合、導電性触媒は白金、炭素等の腐食に強い材料を用いることが好ましい。
【0087】
(封止材)
本発明における光電変換素子は、前記半導体電極と前記対電極との間に前記電解質層を封止する封止材150(図1)を設けてもよい。封止材としては、耐候性、耐光性、高防湿性、耐熱性等を有するものが求められ、電解液の蒸散を防止するために、電解液に不溶な物質が好ましい。具体的には、フィルムに加工したポリエチレン、エチレンビニルアセテート等の樹脂が挙げられる。これらの封止材を電極周辺に張り合わせ、加熱又は圧力を加えながら加熱することによって融着させ、さらに、その周囲をエポキシ系樹脂、シリコン系樹脂等を用いて接着することで、電解液の蒸散をより効果的に防ぐことができる。
【0088】
本発明における光電変換素子は、色素増感太陽電池としてだけでなく、光センサーとしても用いることができる。
【0089】
通常、ラジカルを持つ有機化合物は極めて不安定であり、例えば蒸着技術のように加熱等の物理エネルギーを外界からかけると容易に壊れてしまう。したがって、これまで有機ラジカル化合物を用いて素子を作ること自体が困難とされていた。しかしながら、本発明のπ共役型有機ラジカル化合物においては、その優れた安定性のために、真空蒸着法、イオン化蒸着法、スパッタリング法、プラズマ法といった製膜方法が可能となった。これにより全く新規な用途開発が可能となり、産業的、民生的な分野への応用が期待されている。例を挙げれば、有機エレクトロルミネッセンス素子、有機太陽電池、有機トランジスタ、有機メモリー、量子コンピューター等の有機デバイスである。
【0090】
以上、本発明について説明したが、本発明は上記に限定されるものではない。本発明は、その要旨を逸脱しない範囲で様々な変形が可能である。
【実施例】
【0091】
以下、実施例によって本発明を更に詳細に説明するが、本発明はこれらの実施例に限定されるものではない。
【0092】
[実施例1]
(1)2−フェニルキノリンから2−フェニルキノリン−1−オキシドへの誘導
m−クロロ過安息香酸(東京化成工業(株)製 純度65%)1.2g(4.5mmol)を20mlのクロロホルムに溶解した溶液を氷冷し、同じく氷冷した2−フェニルキノリン(アルドリッチ社製)1.0g(4.9mmol)を含む100mlのクロロホルムにゆっくり加え24時間攪拌した。さらに、m−クロロ過安息香酸(東京化成工業(株)製、純度65%)1.2g(4.5mmol)を溶解したクロロホルム溶液20mlを加え24時間攪拌した。その後、1M NaOH水溶液を100ml加え中和した。数回水洗したのちクロロホルム相を回収し、無水硫酸ナトリウムで乾燥させた。溶媒を除去後、得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 酢酸エチル:n−ヘキサン=1:2)で精製し薄黄色固体の目的物0.78g(収率73%)を得た。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0093】
(2)2−フェニルキノリン−1−オキシドから2,2−ジフェニル−1,2−ジヒドロキノリン−1−オキシルへの誘導
2−フェニルキノリン−1−オキシド500mg(2.3mmol)を3口フラスコに入れてテトラヒドロフラン(関東化学(株)製、有機合成用)40mlに溶解させ、窒素置換した。これに室温でフェニルマグネシウムブロミド(関東化学(株)製、1.0mol/lのテトラヒドロフラン溶液)4.5mlをシリンジで滴下し、2時間攪拌後、飽和塩化アンモニウム水溶液を加え中和した。これをクロロホルム100mlに溶解し、12時間暗所で静置した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム:n−ヘキサン=1:2)で精製し、赤褐色液体350mg(収率52%)の目的物を得た。質量スペクトル、ESRにて目的物の生成を確認した。合成を繰り返し、光電変換素子の作製に必要な量を確保した。
【0094】
(3)有機色素を用いた光電変換素子の作製
ハイビスカスパウダー((株)生活の木製)30mgを脱水エタノール(関東化学(株)製、有機合成用)20mlに溶解させ、飽和色素溶液を調製した。半導体電極として、電極基材、透明電極層、及び半導体層からなる二酸化チタン焼付済電極(西野田電工(株)製、商品名:B−Y1)を準備した。前記半導体電極の半導体層を中心部1cm×1cmの範囲を残して削り取った後、前記飽和色素溶液中に浸漬して24時間静置し、半導体層に色素を吸着させた。その後、室温にて1時間真空乾燥させて半導体電極を作製した。
【0095】
次に、アセトニトリル(関東化学(株)製、特級)10gに、2,2−ジフェニル−1,2−ジヒドロキノリン−1−オキシル0.46g、ビス(トリフルオロメタンスルホニル)イミドリチウム(和光純薬工業(株)製)4.4g、ニトロシルテトラフルオロボレート(アルドリッチ社製)18mg、N−(n−ブチル)ベンズイミダゾール(D. Kuang, C. Klein, S. Ito, J.-E. Moser, R. Humphry-Baker, N. Evans, F. Duriaux, C. Gratzel, S. M. Zakeeruddin, M. Gratzel, Adv. Mater. 19, 1133-1137 (2007)に従って合成)1gを加え、電解液(a)を作製した。
【0096】
半導体層の周囲にパラフィルム(登録商標、エル・エム・エス社製)を巻いた後、半導体層に電解液(a)を20μl滴下し、十分浸みこませた。その後、白金触媒層を具備した対極触媒付きフィルム(ペクセル・テクノロジーズ(株)製、PECF−CAT)を対電極として対向させ、治具にて固定し、光電変換素子を作製した。
【0097】
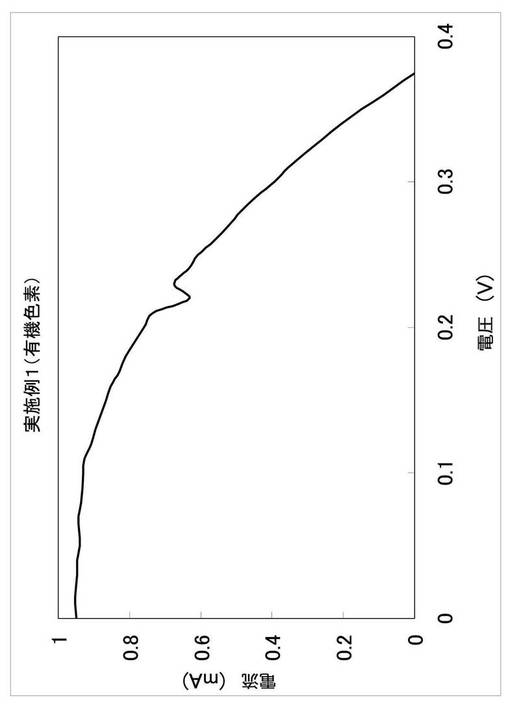
この光電変換素子をペクセル・テクノロジーズ(株)製色素増感太陽電池評価システムを用いて評価した。半導体電極と対電極をI−V特性計測装置PECK2400−N2に接続し、簡易型ソーラーシミュレーターPEC−L01を用いて半導体電極側から光を照射し、AM1.5、100mW/cm2照射条件下における電流(I)−電圧(V)測定を行った。得られたI−V曲線を図2に示す。また、I−V曲線の短絡電流(ISC)、開放電圧(VOC)、フィルファクター(FF)及び光電変換効率(η)を、後述の参考例の結果とともに表1に示す。なお、光電変換効率は以下のように算出された。
【0098】
η(単位:%)=Pout/Pin=100×FF×(ISC×VOC)/(L×A)
ここで、FF=(Imax×Vmax)/(ISC×VOC)
FF=フィルファクター
ISC(単位:mA)=短絡電流
VOC(単位:V)=開放電圧
L(単位:mW/cm2)=照射強度=100mW/cm2
A(単位:cm2)=活性領域=1cm2
Imax(単位:mA)=最大電力点の電流
Vmax(単位:V)=最大電力点の電圧
【0099】
【表1】
【0100】
[実施例2]
金属錯体色素を用いた光電変換素子の作製
色素としてシス−ビス(イソチオシアナト)ビス(2,2’−ビピリジル−4,4’−ジカルボキシラト)ルテニウム(II)(アルドリッチ社製)を用い、色素溶液の濃度を0.3mmol/lとした以外は、実施例1(3)と同様にして光電変換素子を作製し評価を行った。得られたI−V曲線を図3に示す。また、I−V曲線の短絡電流(ISC)、開放電圧(VOC)、フィルファクター(FF)及び光電変換効率(η)を、後述の比較例1の結果とともに表2に示す。
【0101】
【表2】
【0102】
[参考例]
有機色素を用いた光電変換素子の作製
アセトニトリル(関東化学(株)製、特級)10gに、TEMPO(アルドリッチ社製)2g、ビス(トリフルオロメタンスルホニル)イミドリチウム(和光純薬工業(株)製)4.4g、ニトロシルテトラフルオロボレート(アルドリッチ社製)150mg、N−(n−ブチル)ベンズイミダゾール(D. Kuang, C. Klein, S. Ito, J.-E. Moser, R. Humphry-Baker, N. Evans, F. Duriaux, C. Gratzel, S. M. Zakeeruddin, M. Gratzel, Adv. Mater. 19, 1133-1137 (2007)に従って合成)1gを加え、電解液(b)を作製した。
電解液(a)に代えて電解液(b)を用いた以外は、実施例1(3)と同様にして光電変換素子を作製し評価を行った。得られたI−V曲線を図4に示す。また、I−V曲線の短絡電流(ISC)、開放電圧(VOC)、フィルファクター(FF)及び光電変換効率(η)を表1に示す。
【0103】
[比較例1]
金属錯体色素を用いた光電変換素子の作製
電解液(a)に代えて電解液(b)を用いた以外は、実施例2と同様にして光電変換素子を作製し評価を行った。得られたI−V曲線を図5に示す。また、I−V曲線の短絡電流(ISC)、開放電圧(VOC)、フィルファクター(FF)及び光電変換効率(η)を表2に示す。有機色素の場合と異なり、金属錯体色素の場合には光電変換素子としてほとんど機能しない結果となった。
【0104】
[実施例3]
(1)4−クロロアニリンからベンジリデン−(4−クロロアニリン)への誘導
4−クロロアニリン(東京化成工業(株)製)12.7g(0.10mol)を60mlのメタノール(純正化学(株)製、特級)に溶解させた。これに蒸留したベンズアルデヒド(東京化成工業(株)製)10.9g(0.103mol)を添加し、30分間還流しその後放冷した。析出した粗結晶を濾別し、n−ヘキサンで洗浄した。白色固体として目的物20.6gを得た(収率96%)。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0105】
(2)ベンジリデン−(4−クロロアニリン)から2−フェニル−4−エトキシ−6−クロロ−1,2,3,4−テトラヒドロキノリンへの誘導
ベンジリデン−(4−クロロアニリン)2.15g(0.01mol)を100mlのアセトニトリル(関東化学(株)製、特級)に溶解させた。蒸留したエチルビニルエーテル(東京化成工業(株)製、1級)2.16g(0.03mol)と硝酸二アンモニウムセリウム(関東化学(株)製、特級)0.28g(0.5mmol)を加え室温で一時間攪拌した。その後、ジクロロメタンで抽出した。溶媒を除去後、シリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム:n−ヘキサン=1:1)で精製し、緑色の液体1.0gを得た(収率35%)。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0106】
(3)2−フェニル−4−エトキシ−6−クロロ−1,2,3,4−テトラヒドロキノリンから2−フェニル−6−クロロキノリンへの誘導
ナスフラスコに2−フェニル−4−エトキシ−6−クロロ−1,2,3,4−テトラヒドロキノリン2.07g(0.01mol)とトルエン(関東化学(株)製、特級)60mlを加え溶解させた。室温でジエチルアゾジカルボキシレート(東京化成工業(株)製、40%トルエン溶液)6ml(23.2mmol)を加え24時間還流した。水酸化ナトリウム溶液で中和し、分液の後有機相を回収した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム)で精製し、ベージュ色の固体1.80g(収率75%)を得た。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0107】
(4)2−フェニル−6−クロロキノリンから2−フェニル−6−クロロキノリン−1−オキシドへの誘導
m−クロロ過安息香酸(東京化成工業(株)製、純度65%)1.12g(4.2mmol)を20mlのクロロホルムに溶解した溶液を氷冷し、同じく氷冷した2−フェニル−6−クロロキノリン0.96g(4.0mmol)を含む100mlのクロロホルムに滴下し24時間攪拌した。さらに、m−クロロ過安息香酸1.12g(4.2mmol)を溶解したクロロホルム溶液20mlを加え24時間攪拌した。その後で1N水酸化ナトリウム水溶液を100ml加え中和した。3回水洗したのちクロロホルム相を回収し、無水硫酸ナトリウムで乾燥させた。溶媒を除去後、得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム)で精製し薄黄色固体の目的物0.86g(収率79%)を得た。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0108】
(5)2−フェニル−6−クロロキノリン−1−オキシドから2,2−ジフェニル−6−クロロ−1,2−ジヒドロキノリン−1−オキシルへの誘導
2−フェニル−6−クロロキノリン−1−オキシド77mg(0.3mmol)を3口フラスコに入れ5mlのテトラヒドロフラン(関東化学(株)製、有機合成用)に溶解させ、窒素置換した。これに室温でフェニルマグネシウムブロミド(関東化学(株)製、1mol/lテトラヒドロフラン溶液)1.0mlをシリンジで滴下し、2時間攪拌した。反応後、飽和塩化アンモニウム水溶液を加え中和した。これを100mlのクロロホルムに溶解し、12時間暗所で静置した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム:n−ヘキサン=2:1)で精製し、赤褐色液体40mg(収率40%)の目的物を得た。質量スペクトル、元素分析、ESRにて目的物の生成を確認した。合成を繰り返し、光電変換素子の作製に必要な量を確保した。
【0109】
(6)金属錯体色素を用いた光電変換素子の作製
アセトニトリル(関東化学(株)製、特級)10gに、2,2−ジフェニル−6−クロロ−1,2−ジヒドロキノリン−1−オキシル6.4g、ビス(トリフルオロメタンスルホニル)イミドリチウム(和光純薬工業(株)製)4.4g、ニトロシルテトラフルオロボレート(アルドリッチ社製)225mg、N−(n−ブチル)ベンズイミダゾール(D. Kuang, C. Klein, S. Ito, J.-E. Moser, R. Humphry-Baker, N. Evans, F. Duriaux, C. Gratzel, S. M. Zakeeruddin, M. Gratzel, Adv. Mater. 19, 1133-1137 (2007)に従って合成)1gを加え、電解液(c)を作製した。
【0110】
電解液(a)に代えて電解液(c)を用いた以外は、実施例2と同様にして光電変換素子を作製し評価を行った。得られたI−V曲線を図6に示す。また、I−V曲線の短絡電流(ISC)、開放電圧(VOC)、フィルファクター(FF)及び光電変換効率(η)を表3に示す。
【0111】
[実施例4]
(1)2−フェニルキノリン−1−オキシドから2−フェニル−2−(4−クロロフェニル)−1,2−ジヒドロキノリン−1−オキシルへの誘導
実施例1(1)と同様にして得られた2−フェニルキノリン−1−オキシド500mg(2.3mmol)を3口フラスコに入れてテトラヒドロフラン(関東化学(株)製、有機合成用)40mlに溶解させ、窒素置換した。これに室温で4−クロロフェニルマグネシウムブロミド(アルドリッチ社製、1.0mol/lのジエチルエーテル溶液)4.5mlをシリンジで滴下し、2時間攪拌後、飽和塩化アンモニウム水溶液を加え中和した。これをクロロホルム100mlに溶解し、12時間暗所で静置した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム:n−ヘキサン=1:2)で精製し、赤褐色液体244mg(収率32%)の目的物を得た。質量スペクトル、ESRにて目的物の生成を確認した。合成を繰り返し、光電変換素子の作製に必要な量を確保した。
【0112】
(2)金属錯体色素を用いた光電変換素子の作製
アセトニトリル(関東化学(株)製、特級)10gに、2−フェニル−2−(4−クロロフェニル)−1,2−ジヒドロキノリン−1−オキシル6.4g、ビス(トリフルオロメタンスルホニル)イミドリチウム(和光純薬工業(株)製)4.4g、ニトロシルテトラフルオロボレート(アルドリッチ社製)225mg、N−(n−ブチル)ベンズイミダゾール(D. Kuang, C. Klein, S. Ito, J.-E. Moser, R. Humphry-Baker, N. Evans, F. Duriaux, C. Gratzel, S. M. Zakeeruddin, M. Gratzel, Adv. Mater. 19, 1133-1137 (2007)に従って合成)1gを加え、電解液(d)を作製した。
【0113】
電解液(a)に代えて電解液(d)を用いた以外は、実施例2と同様にして光電変換素子を作製し評価を行った。得られたI−V曲線を図7に示す。また、I−V曲線の短絡電流(ISC)、開放電圧(VOC)、フィルファクター(FF)及び光電変換効率(η)を表3に示す。
【0114】
【表3】
【0115】
[実施例5]
(1)6−メトキシキノリンから6−メトキシキノリン−1−オキシドへの誘導
6−メトキシキノリン(東京化成工業 (株)製)2.25g(14.13mmol)を200mlのクロロホルムに溶解させた。ここへ、m−クロロ過安息香酸(東京化成工業(株)製、純度65%)2.49g(9.41mmol)を40mlのクロロホルムに溶解させたもの全量を加え、12時間攪拌した。さらに、m−クロロ過安息香酸2.49g(9.41mmol)を40mlのクロロホルムに溶解させたものを加え、12時間攪拌を続けた。その後、1N水酸化ナトリウム水溶液を250ml加え中和した。これに50mlのクロロホルムを加えて抽出した。クロロホルム相を回収し、無水硫酸ナトリウムで乾燥させた。溶媒を除去し、淡黄色固体の目的物2.2g(収率88%)を得た。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0116】
(2)6−メトキシキノリン−1−オキシドから2−フェニル−6−メトキシキノリン−1−オキシドへの誘導
脱水した6−メトキシキノリン−1−オキシド777mg(4.43mmol)を2口ナスフラスコに入れ、窒素置換した。そこへ、脱水テトラヒドロフラン(関東化学(株)製、有機合成用)50mlに溶解させ、これに室温でフェニルマグネシウムブロミド(関東化学(株)製、1.06mol/lテトラヒドロフラン溶液)8.36mlをシリンジで滴下した。反応終了後、飽和塩化アンモニウム水溶液を加えて中和し、クロロホルムにより抽出を行った。3回水洗したのちクロロホルム相を回収し、無水硫酸ナトリウムで乾燥させた。溶媒を除去後、得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 酢酸エチル:ヘキサン=1:2)で精製し淡黄色固体の目的物710mg(収率64%)を得た。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0117】
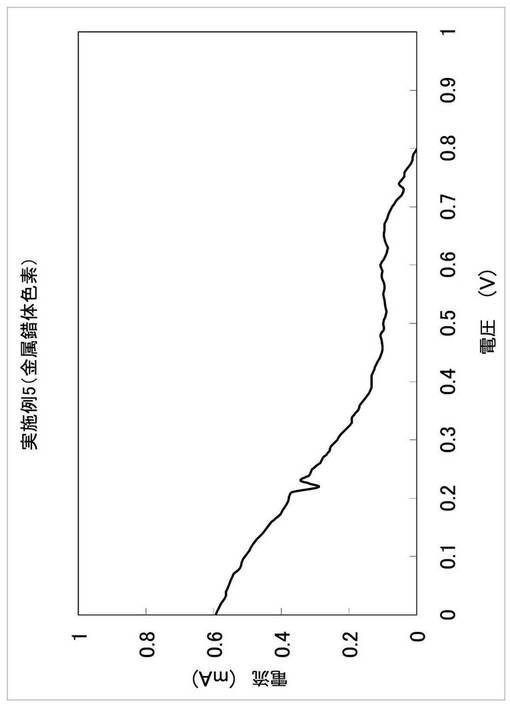
(3)2−フェニル−6−メトキシキノリン−1−オキシドから2,2−ジフェニル−1,2−ジヒドロキノリン−1−オキシルへの誘導
脱水した2−フェニル−6−メトキシキノリン−1−オキシド300mg(1.19mmol)を2口ナスフラスコに入れ、窒素置換した。そこへ、脱水テトラヒドロフラン(関東化学(株)製、有機合成用)30mlに溶解させ、これに室温でフェニルマグネシウムブロミド(関東化学(株)製、1.06mol/lテトラヒドロフラン溶液)1.68mlをシリンジで滴下した。反応終了後、飽和塩化アンモニウム水溶液を加えて中和し、クロロホルムにより抽出を行った。無水硫酸ナトリウムにて脱水後、エバポレーターにて溶媒を除去した。次に、これをクロロホルム(特級)に溶解させ、24時間室温で攪拌した。その後、中性アルミナカラムクロマトグラフィー(展開溶媒 クロロホルム)で精製し、赤褐色固体の目的物137mg(収率35%)を得た。質量スペクトル、ESRにて目的物の生成を確認した。合成を繰り返し、光電変換素子の作製に必要な量を確保した。
【0118】
(4)金属錯体色素を用いた光電変換素子の作製
FTOガラスLow−E(日本板硝子(株)製)を、3cm×3cmの大きさにダイヤモンドカッターで切り、2−プロパノール(和光純薬工業(株)製、特級)に入れた後、超音波洗浄を5分間行った。超音波洗浄機から取り出して乾燥後、40mM塩化チタン水溶液(和光純薬工業(株)製の塩化チタン(IV)溶液を希釈して使用)に60℃で30分間浸漬した。表面の液体をふき取った後、真空乾燥を1時間行った。ドクターブレード法により、ガラス基板の導電面に厚み15μm設定で二酸化チタンPST−18NR(日揮触媒化成(株)製)を塗布し、5分間室温で放置した。塗布されたガラス基板を125℃のホットプレートに6分間置き、325℃に5分間、375℃に5分間、450℃に15分、500℃に15分間と段階的に温度を上げて焼成した。なお、焼成初期段階で色の変化が認められたが、温度を上げることで透明になった。室温になるまで放置した後、厚み20μm設定で二酸化チタンPST−400C(日揮触媒化成(株)製)をさらに塗布し、5分間室温で放置した。塗布されたガラス基板を125℃のホットプレートに6分間置き、325℃に5分間、375℃に5分間、450℃に15分、500℃に15分間と段階的に温度を上げて焼成した。その後、40mM塩化チタン水溶液(和光純薬工業(株)製の塩化チタン(IV)溶液を希釈して使用)に60℃で30分間浸漬した。表面の液体をふき取った後、真空乾燥を1時間行った。室温になるまで放置した。半導体層を中心部1cm×1cmの範囲を残して削り取った後、再びホットプレートにのせて500℃まで昇温し、30分静置した。その間、シス−ビス(イソチオシアナト)ビス(2,2’−ビピリジル−4,4’−ジカルボキシラト)ルテニウム(II)(アルドリッチ社製)を脱水エタノール(関東化学(株)製、有機合成用)に溶解させ、色素溶液の濃度を0.2mmol/lに調製した。ホットプレートの加熱を止めて80℃になった段階で前記色素溶液中に浸漬して24時間静置し、半導体層に色素を吸着させた。その後、室温にて1時間真空乾燥させて半導体電極を作製した。
【0119】
次に、2箇所に直径1mmの穴をあけたFTO硝子(西野田電工(株)製)を2−プロパノール(和光純薬工業(株)製、特級)に入れた後、超音波洗浄を5分間行った。表面の液体をふき取った後、真空乾燥を1時間行った。ドクターブレード法で、透明ガラス電極作製用白金ペーストPlatisol T/SP(Solaronix社製)をスコッチテープ(3M社製)二枚分に相当する厚みで塗布した。40分間室温で放置し、これをホットプレートに載せ350℃で5分間、450℃で15分間焼成して、対電極を得た。1cm×1cmの穴を開けた熱圧着用封止剤Meltonix 1170−25 Series(Solaronix社製)を用いて半導体電極と対極を140℃のアイロンで接着した。
【0120】
次に、アセトニトリル(関東化学(株)製、特級)に対し、2,2−ジフェニル−6−メトキシ−1,2−ジヒドロキノリン−1−オキシル0.1mol/L、ビス(トリフルオロメタンスルホニル)イミドリチウム(和光純薬工業(株)製)1.2mol/L、ニトロシルテトラフルオロボレート(アルドリッチ社製)0.01mol/L、N−(n−ブチル)ベンズイミダゾール(D. Kuang, C. Klein, S. Ito, J.-E. Moser, R. Humphry-Baker, N. Evans, F. Duriaux, C. Gratzel, S. M. Zakeeruddin, M. Gratzel, Adv. Mater. 19, 1133-1137 (2007)に従って合成)0.5mol/Lとなるようにそれぞれ加え、電解液(e)を作製した。調製した電解液(e)を対極の穴からマイクロシリンジで注入し、穴を接着テープで封じることにより光電変換素子を作製した。
【0121】
この光電変換素子を用いた以外は、実施例1(3)と同様にして評価を行った。得られたI−V曲線を図8に示す。また、I−V曲線の短絡電流(ISC)、開放電圧(VOC)、フィルファクター(FF)及び光電変換効率(η)を、表4に示す。
【0122】
[実施例6]
(1)4−ヨードアニリンからベンジリデン−(4−ヨードアニリン)への誘導
4−ヨードアニリン (東京化成工業(株)製、1級)8.0g(37mmol)を50mlのトルエン(関東化学(株)製、特級)に溶解させた。これに蒸留したベンズアルデヒド(東京化成工業(株)製)3.87g(90.5mmol)を添加し、30分間還流しその後放冷した。n−ヘキサンを加えて析出した粗結晶を濾別し、n−ヘキサンでさらに洗浄した。黄色固体として目的物11.0gを得た(収率98%)。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0123】
(2)ベンジリデン−(4−ヨードアニリン)から2−フェニル−4−エトキシ−6−ヨード−1,2,3,4−テトラヒドロキノリンへの誘導
ベンジリデン−(4−ヨードアニリン)3.0g(9.8mmol)を15mlのトルエン(関東化学(株)製、特級)に溶解させた。蒸留したエチルビニルエーテル(東京化成工業(株)製、1級)1.88mL(19.5mmol)とボロントリフルオリドエタレート(関東化学(株)製)41μL(0.33mmol)を加え二時間還流した。その後、室温に戻して10重量%水酸化ナトリウム水溶液で中和し、クロロホルムを加え、分液の後、有機相を回収した。溶媒を除去後、シリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム)で精製し、黄褐色の液体1.9gを得た(収率51%)。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0124】
(3)2−フェニル−4−エトキシ−6−ヨード−1,2,3,4−テトラヒドロキノリンから2−フェニル−6−ヨードキノリンへの誘導
ナスフラスコに2−フェニル−4−エトキシ−6−ヨード−1,2,3,4−テトラヒドロキノリン1.9g(2.5mmol)とトルエン(関東化学(株)製、特級)10mlを加え溶解させた。室温でジエチルアゾジカルボキシレート(東京化成工業(株)製、40%トルエン溶液)2.2ml(5mmol)を加え12時間還流した。水酸化カリウム水溶液で中和し、クロロホルム分液の後有機相を回収した。無水硫酸ナトリウムによる脱水を行った後、溶媒を除去し、得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム)で精製し、薄黄色の固体1.1g(収率65%)を得た。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0125】
(4)2−フェニル−6−ヨードキノリンから2−フェニル−6−(2,2,2−トリフルオロエトキシ)キノリンへの誘導
2−フェニル−6−ヨードキノリン500mg(1.5mmol)に、トリフルオロエタノール(和光純薬工業(株)製)30mL(420mmol)を加え、さらにヨウ化銅(関東化学(株)製)150mg(0.79mmol)、炭酸セシウム(関東化学(株)製、特級)4.2g(12mmol)、及び2−オキソシクロヘキサカルボン酸エチル(東京化成工業(株)製)250μL(1.57mmol)を加えて14時間還流した。ジクロロメタンと水を加え、分液の後、有機相を回収した。無水硫酸ナトリウムによる脱水を行った後、溶媒を除去し、得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム:n−ヘキサン=5:1)で精製し、白色固体210mg(収率57%)を得た。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0126】
(5)2−フェニル−6−(2,2,2−トリフルオロエトキシ)キノリンから2−フェニル−6−(2,2,2−トリフルオロエトキシ)キノリン−1−オキシドへの誘導
m−クロロ過安息香酸(東京化成工業(株)製、純度65%)90mg(0.3mmol)を2mlのクロロホルムに溶解した溶液を氷冷し、同じく氷冷した2−フェニル−6−(2,2,2−トリフルオロエトキシ)キノリン100mg(0.3mmol)を含む10mlのクロロホルムに滴下し24時間攪拌した。さらに、15時間後にm−クロロ過安息香酸90mg(0.3mmol)を溶解したクロロホルム溶液2mlを加え30時間攪拌した。その後、1N水酸化ナトリウム水溶液を10ml加え中和した。3回水洗したのちクロロホルム相を回収し、無水硫酸ナトリウムで乾燥させた。溶媒を除去後、得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム)で精製し黄白色固体の目的物93mg(収率88%)を得た。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0127】
(6)2−フェニル−6−(2,2,2−トリフルオロエトキシ)キノリン−1−オキシドから2,2−ジフェニル−6−(2,2,2−トリフルオロエトキシ)−1,2−ジヒドロキノリン−1−オキシルへの誘導
五酸化二リン(関東化学(株)製、1級)で脱水した2−フェニル−6−(2,2,2−トリフルオロエトキシ)キノリン−1−オキシド80mg(0.25mmol)を2口ナスフラスコに入れ、窒素置換した。脱水テトラヒドロフラン(関東化学(株)製、有機合成用)5mlに溶解させ、これに室温でフェニルマグネシウムブロミド(関東化学(株)製、1mol/lテトラヒドロフラン溶液)0.5mlをシリンジで滴下し、1時間攪拌した。反応後、飽和塩化アンモニウム水溶液を加え中和した。これを100mlのクロロホルムに溶解し、12時間暗所で静置した。無水硫酸ナトリウムで乾燥させ、溶媒を除去後、得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム)で精製し、光沢のある赤褐色固体49mg(収率49%)の目的物を得た。質量スペクトル、元素分析、ESRにて目的物の生成を確認した。合成を繰り返し、光電変換素子の作製に必要な量を確保した。
【0128】
次に、アセトニトリル(関東化学(株)製、特級)に対し、2,2−ジフェニル−6−(2,2,2−トリフルオロエトキシ)−1,2−ジヒドロキノリン−1−オキシル0.1mol/L、ビス(トリフルオロメタンスルホニル)イミドリチウム(和光純薬工業(株)製)1.2mol/L、ニトロシルテトラフルオロボレート(アルドリッチ社製)0.01mol/L、N−(n−ブチル)ベンズイミダゾール(D. Kuang, C. Klein, S. Ito, J.-E. Moser, R. Humphry-Baker, N. Evans, F. Duriaux, C. Gratzel, S. M. Zakeeruddin, M. Gratzel, Adv. Mater. 19, 1133-1137 (2007)に従って合成)0.5mol/Lとなるようにそれぞれ加え、電解液(f)を作製した。
電解液(e)に代えて電解液(f)を用いた以外は、実施例5と同様にして光電変換素子を作製し評価を行った。得られたI−V曲線を図9に示す。また、I−V曲線の短絡電流(ISC)、開放電圧(VOC)、フィルファクター(FF)及び光電変換効率(η)を表4に示す。
【0129】
【表4】
【産業上の利用可能性】
【0130】
本発明によって、電解質層の腐食性が低く、かつ、光電変換効率に優れる光電変換素子及びそれを構成するπ共役型有機ラジカル化合物を提供することができる。また、本発明のπ共役型安定有機ラジカル化合物は、様々な有機デバイスへの適用が可能である。例えば有機エレクトロルミネッセンス素子、有機太陽電池、有機トランジスタ、有機メモリー、量子コンピューター等への利用が期待される。
【符号の説明】
【0131】
110 光電変換素子
120 半導体電極
121 半導体層
122 透明導電層
123 電極基材
130 電解質層
140 対電極
150 封止材
【技術分野】
【0001】
本発明は、光電変換素子及びπ共役型有機ラジカル化合物に関する。
【背景技術】
【0002】
無機化合物によって構成された材料、素子、デバイスを凌ぐ分子ナノエレクトロニクス材料の出現に伴い、有機化合物の機能がエレクトロニクス材料の主役として注目されている。有機化合物をエレクトロニクス材料として用いた有機デバイスの例としては、太陽電池や有機エレクトロルミネッセンス素子、電界効果トランジスタ(Field Effect Transistor:FET)等が挙げられる。これらは有機化合物の電気物性及び光物性を利用したデバイスであり、近年めざましい発展を遂げている。
【0003】
それらの発展に付随して、有機ラジカル化合物を利用した新しい技術開発が行われるようになってきた。有機ラジカル化合物は不対電子に由来する特異な電子・磁気特性を示し、特に強磁性が付与された化合物は、例えば、低重力の宇宙空間において使用する軽量磁石、無機の磁性フィラーと比べて比重の小さい磁性塗料、情報の高密度化が可能な記録材料、外部磁石によりコントロール可能なドラッグデリバリーシステム素材等への応用が期待できる。また、DNA塩基と類似の複素環構造を有し、さらにはラジカル酸化能を有しているので、DNA相互作用物質又はDNA切断剤等への応用も期待できる。
【0004】
実用的な有機ラジカルの代表例としては、ニトロキシドラジカルが挙げられる。ニトロキシドラジカルの電子構造は、不対電子が酸素原子上にある構造と窒素原子上にある構造の共鳴によって表される。さらに、ニトロキシドラジカルは酸素原子上の非共有電子対によりルイス塩基性を示すので、金属イオンとの配位能や水素結合のプロトン受容性を有する。このニトロキシドラジカルを利用した有機デバイスも近年盛んに報告されるようになり、特に、光電変換素子として様々な利用方法が提案されている。
【0005】
例えば、非特許文献1、特許文献1及び特許文献2には、腐食性の高い遊離のヨウ素化合物の代わりに2,2,6,6−テトラメチルピペリジン−N−オキシル(以下「TEMPO」ともいう。)又はその誘導体をレドックスメディエーターとして用いた色素増感太陽電池が記載されている。特許文献3には、配位子にラジカル部位を備えた光吸収材料が記載されている。特許文献4及び特許文献5には、ラジカル化合物からなる有機化合物層を電極と電荷輸送層の間に備えた光電変換素子が記載されている。
【0006】
ところで、TEMPOに代表される非共役型有機ラジカル化合物は、半占軌道(Singly Occupied Molecular Orbital:以下「SOMO」ともいう。)がニトロキシド部位に局在化している。そのために、酸化還元反応の可逆安定性には優れるものの、他の物質との相互作用という観点からは必ずしも最適な構造ではなく、光電変換素子の材料として更なる改良が求められている。特に、色素増感太陽電池のレドックスメディエーターとして用いる場合、錯体色素との相互作用において改善の必要性がある。
【0007】
一方、非特許文献2には、π共役型有機ラジカル化合物である2,2−ジフェニル−1,2−ジヒドロキノリン−1−オキシル(以下「DPQN」ともいう。)及びその誘導体が記載されている。しかしながら、従来公知の文献によるとπ共役平面による自己集積機能のみが研究対象となっていた。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2009−76369号公報
【特許文献2】特開2010−205704号公報
【特許文献3】特開2011−6665号公報
【特許文献4】特開2003−100360号公報
【特許文献5】特開2009−21212号公報
【非特許文献】
【0009】
【非特許文献1】Z. Zhang, P. Chen, T. N. Murakami, S. M. Zakeeruddin, M. Gratzel, Adv. Funct. Mater. 18, 341-346 (2008)
【非特許文献2】N. Yoshioka et al., Chem. Phys. Lett. 402, 11-16 (2005)
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は、上記事情に鑑みてなされたものであり、電解質層の腐食性が低く、かつ、光電変換効率に優れる光電変換素子及びそれを構成するπ共役型有機ラジカル化合物を提供することを目的とする。
【課題を解決するための手段】
【0011】
本発明者は、π共役型有機ラジカル化合物は、SOMOがπ共役平面に非局在化して存在しているため、他の物質との相互作用という観点から、光電変換素子として使用した場合には有利に働く可能性があるのではないかと考えた。そして、上記課題を解決するために鋭意研究を重ねた結果、特定のπ共役型有機ラジカル化合物が腐食性の低い新たな光電変換素子の材料として光電変換効率の向上に寄与することを見出し、本発明を完成するに至った。すなわち、本発明は下記のとおりである。
[1]半導体層及び色素を含む半導体電極と、対電極と、該半導体電極と該対電極との間に設けられた電解質層とを含む光電変換素子であって、該電解質層が、下記一般式(1)で示されるπ共役型有機ラジカル化合物を含む、上記光電変換素子。
【0012】
【化1】
(式(1)中、X1、X2、X3、X4、及びX5で表される置換基は、それぞれ独立に電子吸引性基、水素、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、又は炭素数1〜6のフッ素置換アルコキシ基のいずれかである。)
[2]前記一般式(1)中、X1、X2、X3、X4、及びX5で表される置換基のうち少なくとも1つは電子吸引性基であり、残余の置換基は、それぞれ独立に水素、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、又は炭素数1〜6のフッ素置換アルコキシ基のいずれかである[1]記載の光電変換素子。
[3]前記電子吸引性基が、F、Cl、NO2、CN、C6F5、及びCnF2n+1(nは1〜6の整数)からなる群からそれぞれ独立に選ばれた基である[2]記載の光電変換素子。
[4]前記色素が金属錯体色素である[1]〜[3]のいずれか一項記載の光電変換素子。
[5]下記一般式(1)で示されるπ共役型有機ラジカル化合物。
【0013】
【化2】
(式(1)中、X1、X2、X3、X4、及びX5で表される置換基のうち少なくとも1つは電子吸引性基であり、残余の置換基は、それぞれ独立に水素、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、又は炭素数1〜6のフッ素置換アルコキシ基のいずれかである。)
[6]前記電子吸引性基が、F、Cl、NO2、CN、C6F5、及びCnF2n+1(nは1〜6の整数)からなる群からそれぞれ独立して選ばれた基である[5]記載のπ共役型有機ラジカル化合物。
【発明の効果】
【0014】
本発明によると、電解質層の腐食性が低く、かつ、光電変換効率に優れる光電変換素子及びそれを構成するπ共役型有機ラジカル化合物を提供することができる。
【図面の簡単な説明】
【0015】
【図1】本発明の光電変換素子の一例の断面模式図。
【図2】実施例の光電変換素子のI−V曲線。
【図3】実施例の光電変換素子のI−V曲線。
【図4】参考例の光電変換素子のI−V曲線。
【図5】比較例の光電変換素子のI−V曲線。
【図6】実施例の光電変換素子のI−V曲線。
【図7】実施例の光電変換素子のI−V曲線。
【図8】実施例の光電変換素子のI−V曲線。
【図9】実施例の光電変換素子のI−V曲線。
【発明を実施するための形態】
【0016】
以下、本発明の実施態様(以下「本実施態様」ともいう。)について詳細に説明する。
(光電変換素子)
光電変換素子は、光エネルギーを電気エネルギーに変換する素子であり、色素増感太陽電池を包含する用語と定義する。
【0017】
本実施態様の光電変換素子は、半導体層及び色素を含む半導体電極と、対電極と、前記半導体電極と前記対電極との間に設けられた電解質層とを含むものであり、前記電解質層が下記一般式(1)で示されるπ共役型有機ラジカル化合物を含むことを特徴としている。
【0018】
【化3】
(式(1)中、X1、X2、X3、X4、及びX5で表される置換基は、それぞれ独立に電子吸引性基、水素、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、又は炭素数1〜6のフッ素置換アルコキシ基のいずれかである。)
ここで、前記一般式(1)で示されるπ共役型有機ラジカル化合物は、X1、X2、X3、X4、及びX5で表される置換基のうち少なくとも1つは電子吸引性基であり、残余の置換基は、それぞれ独立に水素、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、又は炭素数1〜6のフッ素置換アルコキシ基のいずれかである化合物が好ましい。また、前記電子吸引性基が、F、Cl、NO2、CN、C6F5、及びCnF2n+1(nは1〜6の整数)からなる群からそれぞれ独立に選ばれた基である化合物がより好ましい。
(π共役型有機ラジカル化合物)
一般式(1)で示されるπ共役型有機ラジカル化合物は、ジフェニル骨格の遮蔽効果によってNO部位が保護されるため、室温大気下においても化学的安定性が保たれている。また、ニトロキシドラジカルの窒素原子がπ共役平面の六員環構造上にあるため、酸化還元状態においてもπ共役平面が開環せず、長期安定性にも優れているという特徴がある。
【0019】
置換基として導入したX1、X2、X3、X4、及びX5は2つの役割を担っている。
【0020】
第一の役割は、π共役平面への不対電子密度の広がりを制御することである。π共役型有機ラジカル化合物は、元々SOMOがπ共役平面に拡張しているため、非共役型有機ラジカル化合物と比較して光電変換素子としての使用時における他の物質との相互作用には有利であると考えられる。そこで、X1、X2、X3、X4、又はX5に置換基として電子吸引性基を導入することによりπ共役平面の不対電子密度を広げることが、他の物質との相互作用を一段と高める効果があるので好ましい。なお、電子吸引性基の導入位置はX1、X2、又はX3の位置がより好ましく、X2の位置が特に好ましい。また、前記電子吸引性基が、F、Cl、NO2、CN、C6F5、及びCnF2n+1(nは1〜6の整数)からなる群からそれぞれ独立に選ばれた基であることがより好ましい。
【0021】
第二の役割は、溶媒への溶解性を制御することである。置換基を導入すると分子容が大きくなるため、π共役平面の自己集積機能を低減させる効果がある。言い換えると、結晶性や溶解性等の物理化学的性質を制御することが可能になる。なお、第二の役割の場合には、電子吸引性基に限らず効果が認められるが、導入位置は第一の役割への影響が少ないX4、又はX5の位置が好ましい。
(π共役型有機ラジカル化合物の製造方法)
本実施態様におけるπ共役型有機ラジカル化合物は、下記2通りの反応ルートのいずれか一方により製造することができる。なお、反応式中、X1、X2、X3、X4、及びX5は前記のとおりである。
[反応ルート1]
【0022】
【化4】
[反応ルート2]
【0023】
【化5】
まず、上記一般式(2)で示される反応ルート1の製造方法について説明する。
【0024】
(i)の反応では、氷冷したm−クロロ過安息香酸のクロロホルム溶液を、同じく氷冷したキノリン誘導体(2A)のクロロホルム溶液に滴下し、その後、m−クロロ過安息香酸をアルカリ水溶液によって加水分解する。アルカリ水溶液として特に制限はないが、水酸化ナトリウム水溶液であることが好ましい。分液による水洗を経てクロロホルム相を回収し、脱水剤を加えて乾燥させる。脱水剤として特に制限はないが、無水硫酸ナトリウム又は無水硫酸マグネシウムであることが好ましい。溶媒を除去後、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製して化合物(2B)を得る。シリカゲルカラムクロマトグラフィーの展開溶媒として特に制限はないが、例えば、酢酸エチル:n−ヘキサン=1:1(体積比)とした展開溶媒等が挙げられる。
【0025】
(ii)の反応では、化合物(2B)を脱水テトラヒドロフランに溶解させ、不活性ガス置換する。不活性ガスとして特に制限はないが、アルゴン又は窒素であることが好ましい。これに置換基X4を有する4−置換フェニルマグネシウムブロミドを含むテトラヒドロフラン溶液を滴下して攪拌する。なお、反応温度について特に制限はないが、0〜60℃であることが好ましく、5〜50℃であることがより好ましく、10〜40℃であることがさらに好ましい。この反応は、低温では反応速度が低下し、高温では脱水反応が併発して酸素が取れた2−フェニルキノリン誘導体になるため、反応温度を前記の範囲内に保つことによって反応収率を高めることができる。反応後は、中和剤によって中和する。中和剤として特に制限はないが、飽和塩化アンモニウム水溶液であることが好ましい。分液による水洗を経てクロロホルム相を回収し、脱水剤を加えて乾燥させる。脱水剤として特に制限はないが、無水硫酸ナトリウム又は無水硫酸マグネシウムであることが好ましい。溶媒を除去後トルエンに溶解して沸点還流し、トルエンを除去した後に得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製して化合物(2C)を得る。シリカゲルカラムクロマトグラフィーの展開溶媒として特に制限はないが、例えば、酢酸エチル:n−ヘキサン=1:1(体積比)とした展開溶媒等が挙げられる。
【0026】
(iii)の反応では、化合物(2C)を脱水テトラヒドロフランに溶解させ、不活性ガス置換する。不活性ガスとして特に制限はないが、アルゴン又は窒素であることが好ましい。これに室温で置換基X5を有する4−置換フェニルマグネシウムブロミドを含むテトラヒドロフラン溶液を滴下して攪拌する。なお、反応温度について特に制限はないが、0〜60℃であることが好ましく、5〜50℃であることがより好ましく、10〜40℃であることがさらに好ましい。この反応は、低温では反応速度が低下し、高温では脱水反応が併発して酸素が取れた2−フェニルキノリン誘導体になるため、反応温度を前記の範囲内に保つことによって反応収率を高めることができる。反応後は、中和剤によって中和する。中和剤として特に制限はないが、飽和塩化アンモニウム水溶液であることが好ましい。なお、この状態ではN−OH体(2D)となっていると推定されるが、不安定であるためキャラクタリゼーションは困難である。したがって、(iii)の反応に続けて(iv)の反応を行うことが好ましい。
【0027】
(iv)の反応では、(iii)で合成した化合物(2D)をクロロホルムに溶解して暗所に静置することにより空気酸化させる。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製することによってπ共役型有機ラジカル化合物(2E)を得ることができる。シリカゲルカラムクロマトグラフィーの展開溶媒として特に制限はないが、例えば、クロロホルム等が挙げられる。
【0028】
次に、上記一般式(3)で示される反応ルート2の製造方法について説明する。
【0029】
(v)の反応では、置換基X1及びX2を有するアニリン誘導体(3A)にエタノールを加え、加熱溶解させる。これに置換基X4を有するベンズアルデヒド誘導体を添加して沸点還流する。析出した粗結晶を濾別し、n−ヘキサン洗浄によって精製して化合物(3B)を得る。
【0030】
(vi)の反応では、化合物(3B)にベンゼンを加え溶解させる。蒸留したアルキルビニルエーテルと三フッ化ホウ素ジエチルエーテル錯体を加えて加熱攪拌する。アルキルビニルエーテルとして特に制限はないが、エチルビニルエーテルであることが好ましい。反応温度について特に制限はないが、25〜60℃であることが好ましく、30〜50℃であることがより好ましく、35〜45℃であることがさらに好ましい。攪拌時間は1〜72時間であることが好ましく、5〜48時間であることがより好ましく、10〜24時間であることがさらに好ましい。なお、(vi)の反応は、溶媒にアセトニトリルを用い、硝酸二アンモニウムセリウムを加えて攪拌しても進行する。室温に戻してアルカリ水溶液で中和し、クロロホルム溶液を加え、分液の後、有機相を回収する。アルカリ水溶液として特に制限はないが、水酸化ナトリウム水溶液であることが好ましい。その後、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製して化合物(3C)を得る。シリカゲルカラムクロマトグラフィーの展開溶媒として特に制限はないが、例えば、クロロホルム等が挙げられる。
【0031】
(vii)の反応では、化合物(3C)にトルエンを加え溶解させる。ジエチルアゾジカルボキシレートを加え沸点還流する。アルカリ水溶液で中和し、分液の後、有機相を回収する。アルカリ水溶液として特に制限はないが、水酸化ナトリウム水溶液であることが好ましい。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製して化合物(3D)を得る。シリカゲルカラムクロマトグラフィーの展開溶媒として特に制限はないが、例えば、クロロホルム:n−ヘキサン=1:1(体積比)とした展開溶媒等が挙げられる。なお、X2がI等のハロゲンである場合には、炭素数1〜6のアルコキシ基又は炭素数1〜6のフッ素置換アルコキシ基への置換が可能である。具体的には、例えば、X2がI等のハロゲンである化合物(3D)に、炭素数1〜6のアルコール又は炭素数1〜6のフッ素置換アルコール、ヨウ化銅、炭酸セシウム、及び2−オキソシクロヘキサカルボン酸エチルを加えて沸点還流する。反応時間は1〜72時間であることが好ましく、5〜48時間であることがより好ましく、10〜24時間であることがさらに好ましい。その後、分液による水洗を経て有機相を回収し、脱水剤を加えて乾燥させる。脱水剤として特に制限はないが、無水硫酸ナトリウム又は無水硫酸マグネシウムであることが好ましい。溶媒を除去後に得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製してX2の置換体が得られる。シリカゲルカラムクロマトグラフィーの展開溶媒として特に制限はないが、例えば、クロロホルム:n−ヘキサン=5:1(体積比)とした展開溶媒等が挙げられる。
【0032】
(viii)の反応では、氷冷したm−クロロ過安息香酸のクロロホルム溶液に、同じく氷冷した化合物(3D)のクロロホルム溶液に加え、その後、m−クロロ過安息香酸をアルカリ水溶液によって加水分解する。アルカリ水溶液として特に制限はないが、水酸化ナトリウム水溶液であることが好ましい。分液による水洗を経てクロロホルム相を回収し、脱水剤を加えて乾燥させる。脱水剤として特に制限はないが、無水硫酸ナトリウム又は無水硫酸マグネシウムであることが好ましい。溶媒を除去後、得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製して化合物(3E)を得る。シリカゲルカラムクロマトグラフィーの展開溶媒として特に制限はないが、例えば、酢酸エチル:n−ヘキサン=1:1(体積比)とした展開溶媒等が挙げられる。
【0033】
(ix)の反応では、化合物(3E)を脱水テトラヒドロフランに溶解させ、不活性ガス置換する。不活性ガスとして特に制限はないが、アルゴン又は窒素であることが好ましい。これに室温で置換基X5を有する4−置換フェニルマグネシウムブロミドを含むテトラヒドロフラン溶液を滴下して攪拌する。なお、反応温度について特に制限はないが、0〜60℃であることが好ましく、5〜50℃であることがより好ましく、10〜40℃であることがさらに好ましい。この反応は、低温では反応速度が低下し、高温では脱水反応が併発して酸素が取れた2−フェニルキノリン誘導体になるため、反応温度を前記の範囲内に保つことによって反応収率を高めることができる。反応後は、中和剤によって中和する。中和剤として特に制限はないが、飽和塩化アンモニウム水溶液であることが好ましい。なお、この状態ではN−OH体(3F)となっていると推定されるが、不安定であるためキャラクタリゼーションは困難である。(ix)の反応に続けて(x)の反応に進むことが好ましい。
【0034】
(x)の反応では、化合物(3F)をクロロホルムに溶解して暗所に静置することにより空気酸化させる。得られた粗生成物をシリカゲルカラムクロマトグラフィーで精製することによってπ共役型有機ラジカル化合物(3G)を得ることができる。シリカゲルカラムクロマトグラフィーの展開溶媒として特に制限はないが、例えば、クロロホルム等が挙げられる。
【0035】
本発明におけるπ共役型有機ラジカル化合物の具体例を以下に例示する。
【0036】
【化6】
【0037】
【化7】
【0038】
【化8】
【0039】
【化9】
【0040】
【化10】
【0041】
【化11】
【0042】
【化12】
【0043】
【化13】
【0044】
【化14】
【0045】
(電解質層)
本実施態様(図1)における電解質層130は、前記のπ共役型有機ラジカル化合物と液体状又は固体状の電解質を含む。電解質層100重量部に対し、前記のπ共役型有機ラジカル化合物が1〜50重量部含まれることが好ましく、1.5〜40重量部含まれることがより好ましく、2〜30重量部含まれることが特に好ましい。この範囲にあることによって、光電変換素子の電解質層として必要な電荷輸送機能が確保され、かつ、析出や濃度ムラ等の影響なく光電変換効率の向上を達成することができる。
【0046】
本実施態様における液体状電解質として特に制限はないが、溶媒及び添加剤から構成されることが好ましい。
【0047】
前記溶媒としては、例えば、アセトニトリル、メトキシアセトニトリル、3−メトキシプロピオニトリル、ベンゾニトリル等のニトリル系溶媒;N−メチルピロリドン、N,N−ジメチルホルムアミド、3−メチル−2−オキサゾリジノン等の含窒素溶媒;γ−ブチロラクトン、バレロラクトン等のラクトン系溶媒;エチレンカーボネート、ジエチルカーボネート、ジメチルカーボネート、エチルメチルカーボネート、プロピレンカーボネート等のカーボネート系溶媒;テトラヒドロフラン、ジオキサン、ジエチルエーテル、エチレングリコールジアルキルエーテル等のエーテル系溶媒;メタノール、エタノール、イソプロピルアルコール、ポリプロピレングリコールモノアルキルエーテル等のアルコール系溶媒;ジメチルスルホキシド、スルホラン等の非プロトン性極性溶媒;等の溶媒が挙げられ、これらはそれぞれ単独で、又は2種以上混合して使用することができる。
【0048】
前記溶媒として、イオン液体、即ち溶融塩を用いることもできる。さらに、液体状電解質が常温溶融塩を含んでもよい。イオン液体としては、「Inorg. Chem.」1996, 35, p1168-1178、「Electrochemistry」2002.2, p130-136、特表平9−507334号公報、特開平8−259543号公報等に開示されている公知の電池や太陽電池等において一般的に使用することができるものであれば、特に限定されない。このうち、室温(25℃)より低い融点を有する塩か、又は室温よりも高い融点を有しても、他の溶融塩や溶融塩以外の添加物を溶解させることにより室温で液状化する塩が好ましく用いられる。
【0049】
具体的には、溶融塩のカチオンとしては、アンモニウム、イミダゾリウム、オキサゾリウム、チアゾリウム、ピペリジニウム、ピラゾリウム、イソオキサゾリウム、チアジアゾリウム、オキサジアゾリウム、トリアゾリウム、ピロリジニウム、ピリジニウム、ピリミジニウム、ピリダジニウム、ピラジニウム、トリアジニウム、ホスホニウム、スルホニウム、カルバゾリウム、インドリウム、及びこれらの誘導体が好ましく、アンモニウム、イミダゾリウム、ピリジニウム、ピペリジニウム、ピラゾリウム、スルホニウムが特に好ましい。
【0050】
また、溶融塩のアニオンとしては、AlCl4−、Al2Cl7−等の金属塩化物、PF6−、BF4−、CF3SO3−、N(CF3SO2)2−、CF3COO−等のフッ素含有物、NO3−、CH3COO−、C6H11COO−、CH3OSO3−、CH3OSO2−、CH3SO3−、CH3SO2−、(CH3O)2PO2−等の非フッ素化合物、塩素、臭素、ヨウ素等のハロゲン化物等が挙げられる。
【0051】
溶融塩は、各種文献や公報で公知の方法により合成することができる。4級アンモニウム塩を例に挙げると、第一段階として3級アミンにアルキル化剤としてアルキルハライドを用いてアミンの4級化を行い、第二段階としてハライドアニオンから目的のアニオンへイオン交換を行うという方法を用いることができる。あるいは、3級アミンを目的のアニオンを有する酸と反応させて一段階で目的の化合物を得る方法がある。
【0052】
溶媒としては、イオン伝導度の高いニトリル化合物又は常温溶融塩を用いることがより好ましい。
【0053】
本発明におけるπ共役型有機ラジカル化合物は、光電変換過程における酸化反応によってカチオン状態となる。このカチオン状態を安定化させるために、添加剤として塩を添加することも可能である。
【0054】
前記添加剤として用いる塩は、例えば、カチオンとして、リチウム、ナトリウム、カリウム、セシウム、カルシウム、ニトロソニウム、アンモニウム、テトラアルキルアンモニウム、イミダゾリウム、オキサゾリウム、チアゾリウム、ピペリジニウム、ピラゾリウム、イソオキサゾリウム、チアジアゾリウム、オキサジアゾリウム、トリアゾリウム、ピロリジニウム、ピリジニウム、ピリミジニウム、ピリダジニウム、ピラジニウム、トリアジニウム、ホスホニウム、スルホニウム、カルバゾリウム、インドリウム、及びこれらの誘導体が挙げられ、リチウム、ニトロソニウム、アンモニウム、イミダゾリウム、ピリジニウム、ピペリジニウム、ピラゾリウム、スルホニウムが好ましい。
【0055】
また、アニオンとしては、PF6−、BF4−、CF3SO3−、N(CF3SO2)2−、F(HF)j−(jは整数)、CF3COO−等のフッ素含有物、NO3−、CH3COO−、C6H11COO−、CH3OSO3−、CH3OSO2−、CH3SO3−、CH3SO2−、(CH3O)2PO2−、SbCl6−等の非フッ素化合物、ヨウ素、臭素等のハロゲン化物等が挙げられる。その他、具体的な組合せとして、フェロシアン酸塩−フェリシアン酸塩、フェロセン−フェリシニウムイオン等の金属錯体、ポリ硫化ナトリウム、アルキルチオール−アルキルジスルフィド等の硫黄化合物、ビオロゲン色素、ヒドロキノン−キノン等も挙げられる。これらの塩は、単独の組み合わせであっても混合であってもよい。なお、前記電解質層における前記塩濃度は、0.05〜20mol/lであることが好ましく、0.1〜15mol/lであることがさらに好ましい。この範囲にあることによって、光電変換素子の電解質層として必要な電荷輸送機能が確保され、かつ、析出や濃度ムラ等の影響なく光電変換効率の向上を達成することができる。
【0056】
本発明における固体状電解質として特に制限はないが、例えば、ポリマー添加、オイルゲル化剤添加、微粒子添加、多官能モノマー類を含む重合、ポリマー架橋反応等の手法により液体状電解質をゲル化又は固体化した電解質が挙げられる。ポリマー添加としては、例えば、ポリアクリロニトリル、ポリフッ化ビニリデン等の添加が挙げられる。オイルゲル化剤添加としては、例えば、ジベンジルデン−D−ソルビトール、コレステロール誘導体、アミノ酸誘導体、トランス−(1R,2R)−1,2−シクロヘキサンジアミンのアルキルアミド誘導体、アルキル尿素誘導体、N−オクチル−D−グルコンアミドベンゾエート、双頭型アミノ酸誘導体、4級アンモニウム誘導体等の添加が挙げられる。
【0057】
本発明における電解質層の形成方法として特に制限はなく、例えば、マイクログラビアコーティング、ディップコーティング、スクリーンコーティング、スピンコーティング等を用いることができる。なお、固体状電解質の場合は、例えば、任意の溶媒によって溶液とした後、上記方法を用いて電極に塗工し、任意の温度に加熱して溶媒を蒸発させればよい。
【0058】
(半導体電極)
本実施態様(図1)における半導体電極120は、導電性基板とその上に形成された半導体層121からなり、導電性基板及び半導体層が光電変換素子110の外側から内側に向かってこの順に積層している。また、半導体層121には光増感剤として機能する色素が含まれている。
【0059】
前記導電性基板は、電極基材123と透明導電層122からなり、透明導電層の面が半導体層に接する。電極基材に用いられる材料は、透明であれば特に限定されるものではない。具体的には、ガラスや強化ガラス等のガラス類、ポリカーボネート、ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリエチレンナフタレート、ポリエチレンスルフィド、ポリスチレン、ポリメチルメタクリレート、ポリメタクリレート、アクリル樹脂、ポリ塩化ビニル等、トリアセチルセルロース、ポリイミド、環状ポリオレフィン、ポリエチレン、ポリプロピレン等のポリオレフィン系等のプラスチックフィルムを用いることができる。
【0060】
前記電極基材の厚さは、材料がプラスチックフィルムの場合は、10〜500μmが好ましく、50〜200μmがより好ましく、100〜200μmが特に好ましい。ガラス類の場合は、0.1〜5mmが好ましく、0.5〜4mmがより好ましく、0.5〜2.5mmが特に好ましい。また、電極基材は光の入射面として用いる場合、可視光域の透過率が85%以上であって、耐候性に優れ、かつ指示材として耐えうる強度を持つことが好ましい。このような電極基材は、必要に応じて表面がコロナ処理、プラズマ処理、薬品処理等によって改質されたものであってもよい。
【0061】
前記透明導電層としては、公知の可視光領域の吸収が少なく導電性のある透明導電材料を用いることができる。透明導電材料としては、錫をドープした酸化インジウム(ITO)、亜鉛をドープした酸化インジウム(IZO)、フッ素やインジウムやアンチモン等をドープした酸化錫、アルミニウムやガリウム等をドープした酸化亜鉛等が好ましい。また、銀又は銀合金(AgAuCu等)をITO、TiO2、ZnO等で挟んだ3層型透明導電材料も挙げられる。これらの中でも、特にITO又はフッ素をドープした酸化錫を使用することが好ましい。
【0062】
前記透明導電層の形成方法としては、真空蒸着法、反応性蒸着法、イオンビームアシスト蒸着法、スパッタリング法、イオンプレーティング法、プラズマCVD法等の真空製膜プロセスによることができる。しかしこれらの例に限定されることはなく、いかなる成膜方法であっても構わない。透明導電層の厚さは50nm〜1μmが好ましい。条件にもよるが、ITOの場合は、100〜400nmの膜厚が好ましく、フッ素ドープ酸化錫の場合は、300〜900nmが好ましい。透明導電層は、可視光域の透過率が65%以上であることが好ましい。
【0063】
本実施態様における半導体層を構成する材料としては、n型又はp型半導体の性質を示す金属酸化物等が挙げられ、例えば、亜鉛、ニオブ、錫、チタン、バナジウム、インジウム、タングステン、タンタル、ジルコニウム、モリブデン、マンガン、鉄、銅、ニッケル、イリジウム、ロジウム、クロム、ルテニウムの酸化物等が挙げられる。また、SrTiO3、CaTiO3、BaTiO3、MgTiO3、SrNb2O6のようなペロブスカイト、硫化カドミウム等、又は、これらの複合酸化物若しくは酸化物混合物等も使用することができる。これらの半導体材料は単独で用いることも2種類以上を混合して用いることもできる。
【0064】
これらの中でも、変換効率、安定性、安全性の観点からは、半導体層が、酸化チタンを含む半導体材料により構成されていることが好ましい。酸化チタンとして、さらに具体的には、アナターゼ型酸化チタン、ルチル型酸化チタン、無定形酸化チタン、メタチタン酸、オルソチタン酸等の種々の酸化チタン、含酸化チタン複合体等が挙げられる。その中でも、光電変換の安定性をさらに向上させる観点からは、アナターゼ型酸化チタンであることが好ましい。また、半導体層が、多孔性のチタニアであってもよい。
【0065】
半導体層の形状としては、半導体微粒子等を焼結することにより得られる多孔性半導体層、ゾル−ゲル法・スパッタ法・スプレー熱分解法等により得られる薄膜状半導体層等が挙げられる。また、その他繊維状半導体層や針状晶からなる半導体層としてもよい。半導体層の形状は、光電変換素子の使用目的に応じて、適宜選択することができる。
【0066】
このうち、色素吸着量等の観点からは、多孔性半導体層、針状晶からなる半導体層等比表面積の大きな半導体層が好ましい。さらに、半導体微粒子の粒径により入射光の利用率等を調整できるという観点からは、半導体層として半導体微粒子から形成される多孔性半導体層を用いることが好ましい。
【0067】
また、半導体層は、単層であっても多層であってもよい。多層にすることによって、充分な厚さの半導体層をさらに容易に形成することができる。
【0068】
また、半導体微粒子から形成される多孔性の多層半導体層は、半導体微粒子の平均粒径の異なる複数の半導体層からなってもよい。たとえば、光入射側に近い方の半導体層(第1半導体層)の半導体微粒子の平均粒径を、光入射側から遠い方の半導体層(第2半導体層)より小さくすることにより、第1半導体層で多くの光を吸収させるとともに、第1半導体層を通過した光を第2半導体層で効率よく散乱させて第1半導体層に戻し、第1半導体層で吸収させることにより、全体の光吸収率をより一層向上させることができる。
【0069】
半導体層の膜厚は、特に限定されるものではないが、透過性、変換効率等の観点より、0.5〜45μmが好ましい。半導体層の比表面積は、多量の色素を吸着によって含ませる観点から、10〜200m2/gが好ましい。
【0070】
また、多孔性の半導体層に色素を吸着させた構成として、電解質中のイオンがさらに充分に拡散して電荷輸送が行われるためには、多孔性の半導体層空隙率を40〜80%とすることが好ましい。なお、空隙率とは、半導体層の体積のうち、半導体層中の空隙が占める体積の割合を%で示したものである。
【0071】
次に、前記半導体層の形成方法について、多孔性半導体層を例にとって説明する。多孔性半導体層は、例えば、半導体微粒子を高分子等の有機化合物及び分散剤とともに、有機溶媒や水等の分散媒に加えて懸濁液を調製し、この懸濁液を透明導電層上に塗布し、これを乾燥、焼成することによって形成する。
【0072】
半導体微粒子とともに分散媒に有機化合物を添加しておくと、焼成時に有機化合物が燃焼して多孔性半導体層内にさらに充分な隙間を確保することが可能となる。また焼成時に燃焼する有機化合物の分子量や添加量を制御することで空隙率を変化させることができる。なお、有機化合物の種類や量は、使用する微粒子の状態、懸濁液全体の総重量等により適宜選択し調整することができる。ただし、半導体微粒子の割合が懸濁液全体の総重量に対して10wt%以上のときは、作製した膜の強度をより一層充分に強くすることができ、半導体微粒子の割合が懸濁液全体の総重量に対して40wt%以下であれば、空隙率が大きな多孔性半導体層をより一層安定的に得ることができるため、半導体微粒子の割合は懸濁液全体の総重量に対して10〜40wt%であることが好ましい。
【0073】
前記の半導体微粒子としては、適当な平均粒径、たとえば、1〜500nm程度の平均粒径を有する単一半導体又は化合物半導体の粒子等が用いられる。その中でも比表面積を大きくするという点からは、1〜50nm程度の平均粒径のものが望ましい。また入射光の利用率を高めるために、200〜400nm程度の平均粒径の比較的大きな半導体粒子を添加してもよい。
【0074】
また、半導体微粒子の製造方法としては、水熱合成法等のゾル−ゲル法、硫酸法、塩素法等が挙げられ、目的の微粒子を製造できる方法であればどんな方法を用いてもよいが、結晶性の観点からは、水熱合成法により合成することが好ましい。
【0075】
前記の有機化合物は、懸濁液中に溶解し、焼成するときに燃焼して除去できるものであれば何でも用いることができる。たとえば、ポリエチレングリコール、エチルセルロース等の高分子が挙げられる。懸濁液の分散媒としては、エチレングリコールモノメチルエーテル等のグライム系溶媒;イソプロピルアルコール等のアルコール類;及びイソプロピルアルコール/トルエン等の混合溶媒;並びに水等が挙げられる。
【0076】
懸濁液の塗布方法としては、ドクターブレード法、スキージ法、スピンコート法、スクリーン印刷法等公知の方法が挙げられる。その後、塗膜の乾燥、焼成を行う。乾燥と焼成の条件は、たとえば大気下又は不活性ガス雰囲気下、50〜800℃程度の範囲内で、10秒〜12時間程度とする。この乾燥及び焼成は、単一の温度で1回又は温度を変化させて2回以上行うことができる。
【0077】
なお、ここでは、多孔性半導体層の形成方法について詳述したが、他の種類の半導体層も種々の公知の方法を用いて形成することができる。例えば、金属酸化物の成膜には、形成したい金属酸化物に対応する金属、金属酸化物、金属亜酸化物等を蒸着源として電子ビームやプラズマ銃による加熱を用いた蒸着法、あるいは酸素ガスを導入しながら蒸着を行なう反応性蒸着法を用いることができる。成膜圧力は用いる蒸着源の種類によって異なるが、1×10−2〜1Paの範囲で行うことが好ましい。成膜の際、任意のガスを用いたプラズマやイオン銃、ラジカル銃等でアシストを行ってもよい。基板温度は、−50℃〜600℃の間で任意に選択することができるが、多孔性を高く保つためには300℃以下であることが好ましい。また目的の金属酸化物によっては、スパッタリング法、イオンプレーティング、CVD等の真空成膜法を用いてもよい。また基材にプラスチックフィルムを用いた場合には、ロールトゥロール方式で成膜すればより高い生産性を得ることができる。
【0078】
以上で得られた金属酸化物膜は、プラズマ処理、コロナ処理、UV処理、薬品処理等の任意の方法で表面処理することができる。また、熱による焼成や圧縮機を用いた加圧処置、レーザアニーリング等の任意の手段を用いて後処理することもできる。
【0079】
(色素)
本実施態様において半導体層に含まれる色素は、光増感剤として機能する。具体的には、色素は種々の可視光領域及び赤外光領域に吸収を持つものであって、吸収係数が大きくかつ繰り返しの酸化還元に対して安定であることが好ましい。また、半導体層に強固に吸着させるために、色素分子中にカルボキシル(COOH)基、アルコキシ基、ヒドロキシル基、ヒドロキシアルキル基、スルホン酸基、エステル基、メルカプト基、ホスホニル基、アミド基、アミノ基、カルボニル基、リン酸基等のインターロック基を有するものが好ましい。この中でも、色素を多孔質半導体表面にさらに安定的に吸着させる観点からは、カルボキシル基を有するものが特に好ましい。
【0080】
インターロック基は、色素の励起状態のエネルギー帯と半導体の伝導帯との間の電子移動を容易にする電気的結合を供給するものである。これらインターロック基を含有する色素としては、たとえば、Ru金属錯体色素(ルテニウムビピリジン系金属錯体色素、ルテニウムターピリジン系金属錯体色素、ルテニウムクォーターピリジン系金属錯体色素等)、Os金属錯体色素、Fe金属錯体色素、Cu金属錯体色素、Pt金属錯体色素、Re金属錯体色素等の金属錯体色素、シアニン系色素、メロシアニン系色素、アゾ系色素、キノン系色素、キノンイミン系色素、キナクリドン系色素、スクアリリウム系色素、トリフェニルメタン系色素、キサンテン系色素、ポルフィリン系色素、フタロシアニン系色素、ペリレン系色素、インジゴ系色素、ナフタロシアニン系色素、クマリン系色素、マーキュロクロム系色素、シアニジン系色素、ローダミン系色素等が挙げられる。その中でも、光電変換反応の安定性の観点からは、金属錯体色素が好ましく、Ru金属錯体色素がより好ましい。
【0081】
前記の半導体層に前記の色素を吸着させる方法としては、たとえば導電性基板上に形成された半導体層を、色素を溶解した溶液に浸漬する方法が挙げられる。色素を溶解するために用いる溶媒は、例えば、エタノール等のアルコール類;アセトン等のケトン類;ジエチルエーテル、テトラヒドロフラン等のエーテル類;アセトニトリル等の窒素化合物;クロロホルム等のハロゲン化脂肪族炭化水素;ヘキサン等の脂肪族炭化水素、ベンゼン等の芳香族炭化水素;酢酸エチル等のエステル類等が挙げられる。またこれらの溶媒は2種類以上を混合して用いてもよい。
【0082】
溶液中の色素濃度、使用する色素及び溶媒の種類は適宜調整することができるが、吸着機能を向上させる観点からは、ある程度高濃度である方が好ましく、例えば、5×10−5mol/l以上の濃度であればよい。
【0083】
色素を溶解した溶液中に半導体層を形成した導電性基板を浸漬する際、溶液及び雰囲気の温度及び圧力は特に限定されるものではなく、例えば室温程度、かつ大気圧下が挙げられ、浸漬時間は使用する色素、溶媒の種類、溶液の濃度等により適宜調整することができる。なお、効果的に行うには加熱下にて浸漬を行えばよい。これにより、半導体層に色素をさらに効率よく吸着させることができる。また、色素を吸着する際、色素の吸着状態を制御するため、色素を溶解した溶液にデオキシコール酸やグアニジンチオシアネート、tert−ブチルピリジン、N−メチルベンズイミダゾール、N−(n−ブチル)ベンズイミダゾール、エタノール等の有機化合物を加えてもよい。
【0084】
(対電極)
本実施態様(図1)における対電極140としては、導電性被膜を形成できる程度の平滑性を備え、かつ封止材を挟み込む程度の強度を有するものであれば、特に限定されるものではなく、無機系材料、有機系材料、金属系材料等の材質を問わず用いることができる。具体的には、白金、金、銀等の貴金属材料、銅、アルミニウム、炭素等の導電性材料が挙げられる。腐食や長期耐久性を考慮すると、白金、金、銀等の貴金属材料や炭素が好ましく、さらにこれらの貴金属又は炭素を蒸着したガラス又はプラスチックフィルム等を使用することも経済性の観点から好ましい。また、可視光透過性を有する光電変換素子を得るために、ITO、酸化錫、フッ素ドープされた酸化錫等の透明導電膜を使用することもできる。なお、対電極の厚さは、材質にもよるが、強度と製品の軽量性を考慮して10〜3000μmが好ましく、50〜1000μmがより好ましい。
【0085】
本発明における対電極には、必要に応じて導電性触媒層を設けてもよい。導電性触媒層としては、任意の導電性材料を用いることができ、白金、金、銀、銅等の金属、又は炭素等を挙げることができる。また、ポリアニリン、ポリチオフェン、PEDOT、ポリピロール等の導電性材料を用いることもできる。これらを形成する際には、透明導電層と同様の真空成膜法、あるいはこれら材料の微粒子をペーストにしたものをウェットコーティングする方法を用いることができる。
【0086】
前記導電性触媒層の厚さは、0.1〜2000nmが好ましく、1〜500nmがより好ましく、5〜200nmがさらに好ましい。なお、腐食性の高い遊離のヨウ素化合物等が含まれる場合、導電性触媒は白金、炭素等の腐食に強い材料を用いることが好ましい。
【0087】
(封止材)
本発明における光電変換素子は、前記半導体電極と前記対電極との間に前記電解質層を封止する封止材150(図1)を設けてもよい。封止材としては、耐候性、耐光性、高防湿性、耐熱性等を有するものが求められ、電解液の蒸散を防止するために、電解液に不溶な物質が好ましい。具体的には、フィルムに加工したポリエチレン、エチレンビニルアセテート等の樹脂が挙げられる。これらの封止材を電極周辺に張り合わせ、加熱又は圧力を加えながら加熱することによって融着させ、さらに、その周囲をエポキシ系樹脂、シリコン系樹脂等を用いて接着することで、電解液の蒸散をより効果的に防ぐことができる。
【0088】
本発明における光電変換素子は、色素増感太陽電池としてだけでなく、光センサーとしても用いることができる。
【0089】
通常、ラジカルを持つ有機化合物は極めて不安定であり、例えば蒸着技術のように加熱等の物理エネルギーを外界からかけると容易に壊れてしまう。したがって、これまで有機ラジカル化合物を用いて素子を作ること自体が困難とされていた。しかしながら、本発明のπ共役型有機ラジカル化合物においては、その優れた安定性のために、真空蒸着法、イオン化蒸着法、スパッタリング法、プラズマ法といった製膜方法が可能となった。これにより全く新規な用途開発が可能となり、産業的、民生的な分野への応用が期待されている。例を挙げれば、有機エレクトロルミネッセンス素子、有機太陽電池、有機トランジスタ、有機メモリー、量子コンピューター等の有機デバイスである。
【0090】
以上、本発明について説明したが、本発明は上記に限定されるものではない。本発明は、その要旨を逸脱しない範囲で様々な変形が可能である。
【実施例】
【0091】
以下、実施例によって本発明を更に詳細に説明するが、本発明はこれらの実施例に限定されるものではない。
【0092】
[実施例1]
(1)2−フェニルキノリンから2−フェニルキノリン−1−オキシドへの誘導
m−クロロ過安息香酸(東京化成工業(株)製 純度65%)1.2g(4.5mmol)を20mlのクロロホルムに溶解した溶液を氷冷し、同じく氷冷した2−フェニルキノリン(アルドリッチ社製)1.0g(4.9mmol)を含む100mlのクロロホルムにゆっくり加え24時間攪拌した。さらに、m−クロロ過安息香酸(東京化成工業(株)製、純度65%)1.2g(4.5mmol)を溶解したクロロホルム溶液20mlを加え24時間攪拌した。その後、1M NaOH水溶液を100ml加え中和した。数回水洗したのちクロロホルム相を回収し、無水硫酸ナトリウムで乾燥させた。溶媒を除去後、得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 酢酸エチル:n−ヘキサン=1:2)で精製し薄黄色固体の目的物0.78g(収率73%)を得た。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0093】
(2)2−フェニルキノリン−1−オキシドから2,2−ジフェニル−1,2−ジヒドロキノリン−1−オキシルへの誘導
2−フェニルキノリン−1−オキシド500mg(2.3mmol)を3口フラスコに入れてテトラヒドロフラン(関東化学(株)製、有機合成用)40mlに溶解させ、窒素置換した。これに室温でフェニルマグネシウムブロミド(関東化学(株)製、1.0mol/lのテトラヒドロフラン溶液)4.5mlをシリンジで滴下し、2時間攪拌後、飽和塩化アンモニウム水溶液を加え中和した。これをクロロホルム100mlに溶解し、12時間暗所で静置した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム:n−ヘキサン=1:2)で精製し、赤褐色液体350mg(収率52%)の目的物を得た。質量スペクトル、ESRにて目的物の生成を確認した。合成を繰り返し、光電変換素子の作製に必要な量を確保した。
【0094】
(3)有機色素を用いた光電変換素子の作製
ハイビスカスパウダー((株)生活の木製)30mgを脱水エタノール(関東化学(株)製、有機合成用)20mlに溶解させ、飽和色素溶液を調製した。半導体電極として、電極基材、透明電極層、及び半導体層からなる二酸化チタン焼付済電極(西野田電工(株)製、商品名:B−Y1)を準備した。前記半導体電極の半導体層を中心部1cm×1cmの範囲を残して削り取った後、前記飽和色素溶液中に浸漬して24時間静置し、半導体層に色素を吸着させた。その後、室温にて1時間真空乾燥させて半導体電極を作製した。
【0095】
次に、アセトニトリル(関東化学(株)製、特級)10gに、2,2−ジフェニル−1,2−ジヒドロキノリン−1−オキシル0.46g、ビス(トリフルオロメタンスルホニル)イミドリチウム(和光純薬工業(株)製)4.4g、ニトロシルテトラフルオロボレート(アルドリッチ社製)18mg、N−(n−ブチル)ベンズイミダゾール(D. Kuang, C. Klein, S. Ito, J.-E. Moser, R. Humphry-Baker, N. Evans, F. Duriaux, C. Gratzel, S. M. Zakeeruddin, M. Gratzel, Adv. Mater. 19, 1133-1137 (2007)に従って合成)1gを加え、電解液(a)を作製した。
【0096】
半導体層の周囲にパラフィルム(登録商標、エル・エム・エス社製)を巻いた後、半導体層に電解液(a)を20μl滴下し、十分浸みこませた。その後、白金触媒層を具備した対極触媒付きフィルム(ペクセル・テクノロジーズ(株)製、PECF−CAT)を対電極として対向させ、治具にて固定し、光電変換素子を作製した。
【0097】
この光電変換素子をペクセル・テクノロジーズ(株)製色素増感太陽電池評価システムを用いて評価した。半導体電極と対電極をI−V特性計測装置PECK2400−N2に接続し、簡易型ソーラーシミュレーターPEC−L01を用いて半導体電極側から光を照射し、AM1.5、100mW/cm2照射条件下における電流(I)−電圧(V)測定を行った。得られたI−V曲線を図2に示す。また、I−V曲線の短絡電流(ISC)、開放電圧(VOC)、フィルファクター(FF)及び光電変換効率(η)を、後述の参考例の結果とともに表1に示す。なお、光電変換効率は以下のように算出された。
【0098】
η(単位:%)=Pout/Pin=100×FF×(ISC×VOC)/(L×A)
ここで、FF=(Imax×Vmax)/(ISC×VOC)
FF=フィルファクター
ISC(単位:mA)=短絡電流
VOC(単位:V)=開放電圧
L(単位:mW/cm2)=照射強度=100mW/cm2
A(単位:cm2)=活性領域=1cm2
Imax(単位:mA)=最大電力点の電流
Vmax(単位:V)=最大電力点の電圧
【0099】
【表1】
【0100】
[実施例2]
金属錯体色素を用いた光電変換素子の作製
色素としてシス−ビス(イソチオシアナト)ビス(2,2’−ビピリジル−4,4’−ジカルボキシラト)ルテニウム(II)(アルドリッチ社製)を用い、色素溶液の濃度を0.3mmol/lとした以外は、実施例1(3)と同様にして光電変換素子を作製し評価を行った。得られたI−V曲線を図3に示す。また、I−V曲線の短絡電流(ISC)、開放電圧(VOC)、フィルファクター(FF)及び光電変換効率(η)を、後述の比較例1の結果とともに表2に示す。
【0101】
【表2】
【0102】
[参考例]
有機色素を用いた光電変換素子の作製
アセトニトリル(関東化学(株)製、特級)10gに、TEMPO(アルドリッチ社製)2g、ビス(トリフルオロメタンスルホニル)イミドリチウム(和光純薬工業(株)製)4.4g、ニトロシルテトラフルオロボレート(アルドリッチ社製)150mg、N−(n−ブチル)ベンズイミダゾール(D. Kuang, C. Klein, S. Ito, J.-E. Moser, R. Humphry-Baker, N. Evans, F. Duriaux, C. Gratzel, S. M. Zakeeruddin, M. Gratzel, Adv. Mater. 19, 1133-1137 (2007)に従って合成)1gを加え、電解液(b)を作製した。
電解液(a)に代えて電解液(b)を用いた以外は、実施例1(3)と同様にして光電変換素子を作製し評価を行った。得られたI−V曲線を図4に示す。また、I−V曲線の短絡電流(ISC)、開放電圧(VOC)、フィルファクター(FF)及び光電変換効率(η)を表1に示す。
【0103】
[比較例1]
金属錯体色素を用いた光電変換素子の作製
電解液(a)に代えて電解液(b)を用いた以外は、実施例2と同様にして光電変換素子を作製し評価を行った。得られたI−V曲線を図5に示す。また、I−V曲線の短絡電流(ISC)、開放電圧(VOC)、フィルファクター(FF)及び光電変換効率(η)を表2に示す。有機色素の場合と異なり、金属錯体色素の場合には光電変換素子としてほとんど機能しない結果となった。
【0104】
[実施例3]
(1)4−クロロアニリンからベンジリデン−(4−クロロアニリン)への誘導
4−クロロアニリン(東京化成工業(株)製)12.7g(0.10mol)を60mlのメタノール(純正化学(株)製、特級)に溶解させた。これに蒸留したベンズアルデヒド(東京化成工業(株)製)10.9g(0.103mol)を添加し、30分間還流しその後放冷した。析出した粗結晶を濾別し、n−ヘキサンで洗浄した。白色固体として目的物20.6gを得た(収率96%)。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0105】
(2)ベンジリデン−(4−クロロアニリン)から2−フェニル−4−エトキシ−6−クロロ−1,2,3,4−テトラヒドロキノリンへの誘導
ベンジリデン−(4−クロロアニリン)2.15g(0.01mol)を100mlのアセトニトリル(関東化学(株)製、特級)に溶解させた。蒸留したエチルビニルエーテル(東京化成工業(株)製、1級)2.16g(0.03mol)と硝酸二アンモニウムセリウム(関東化学(株)製、特級)0.28g(0.5mmol)を加え室温で一時間攪拌した。その後、ジクロロメタンで抽出した。溶媒を除去後、シリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム:n−ヘキサン=1:1)で精製し、緑色の液体1.0gを得た(収率35%)。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0106】
(3)2−フェニル−4−エトキシ−6−クロロ−1,2,3,4−テトラヒドロキノリンから2−フェニル−6−クロロキノリンへの誘導
ナスフラスコに2−フェニル−4−エトキシ−6−クロロ−1,2,3,4−テトラヒドロキノリン2.07g(0.01mol)とトルエン(関東化学(株)製、特級)60mlを加え溶解させた。室温でジエチルアゾジカルボキシレート(東京化成工業(株)製、40%トルエン溶液)6ml(23.2mmol)を加え24時間還流した。水酸化ナトリウム溶液で中和し、分液の後有機相を回収した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム)で精製し、ベージュ色の固体1.80g(収率75%)を得た。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0107】
(4)2−フェニル−6−クロロキノリンから2−フェニル−6−クロロキノリン−1−オキシドへの誘導
m−クロロ過安息香酸(東京化成工業(株)製、純度65%)1.12g(4.2mmol)を20mlのクロロホルムに溶解した溶液を氷冷し、同じく氷冷した2−フェニル−6−クロロキノリン0.96g(4.0mmol)を含む100mlのクロロホルムに滴下し24時間攪拌した。さらに、m−クロロ過安息香酸1.12g(4.2mmol)を溶解したクロロホルム溶液20mlを加え24時間攪拌した。その後で1N水酸化ナトリウム水溶液を100ml加え中和した。3回水洗したのちクロロホルム相を回収し、無水硫酸ナトリウムで乾燥させた。溶媒を除去後、得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム)で精製し薄黄色固体の目的物0.86g(収率79%)を得た。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0108】
(5)2−フェニル−6−クロロキノリン−1−オキシドから2,2−ジフェニル−6−クロロ−1,2−ジヒドロキノリン−1−オキシルへの誘導
2−フェニル−6−クロロキノリン−1−オキシド77mg(0.3mmol)を3口フラスコに入れ5mlのテトラヒドロフラン(関東化学(株)製、有機合成用)に溶解させ、窒素置換した。これに室温でフェニルマグネシウムブロミド(関東化学(株)製、1mol/lテトラヒドロフラン溶液)1.0mlをシリンジで滴下し、2時間攪拌した。反応後、飽和塩化アンモニウム水溶液を加え中和した。これを100mlのクロロホルムに溶解し、12時間暗所で静置した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム:n−ヘキサン=2:1)で精製し、赤褐色液体40mg(収率40%)の目的物を得た。質量スペクトル、元素分析、ESRにて目的物の生成を確認した。合成を繰り返し、光電変換素子の作製に必要な量を確保した。
【0109】
(6)金属錯体色素を用いた光電変換素子の作製
アセトニトリル(関東化学(株)製、特級)10gに、2,2−ジフェニル−6−クロロ−1,2−ジヒドロキノリン−1−オキシル6.4g、ビス(トリフルオロメタンスルホニル)イミドリチウム(和光純薬工業(株)製)4.4g、ニトロシルテトラフルオロボレート(アルドリッチ社製)225mg、N−(n−ブチル)ベンズイミダゾール(D. Kuang, C. Klein, S. Ito, J.-E. Moser, R. Humphry-Baker, N. Evans, F. Duriaux, C. Gratzel, S. M. Zakeeruddin, M. Gratzel, Adv. Mater. 19, 1133-1137 (2007)に従って合成)1gを加え、電解液(c)を作製した。
【0110】
電解液(a)に代えて電解液(c)を用いた以外は、実施例2と同様にして光電変換素子を作製し評価を行った。得られたI−V曲線を図6に示す。また、I−V曲線の短絡電流(ISC)、開放電圧(VOC)、フィルファクター(FF)及び光電変換効率(η)を表3に示す。
【0111】
[実施例4]
(1)2−フェニルキノリン−1−オキシドから2−フェニル−2−(4−クロロフェニル)−1,2−ジヒドロキノリン−1−オキシルへの誘導
実施例1(1)と同様にして得られた2−フェニルキノリン−1−オキシド500mg(2.3mmol)を3口フラスコに入れてテトラヒドロフラン(関東化学(株)製、有機合成用)40mlに溶解させ、窒素置換した。これに室温で4−クロロフェニルマグネシウムブロミド(アルドリッチ社製、1.0mol/lのジエチルエーテル溶液)4.5mlをシリンジで滴下し、2時間攪拌後、飽和塩化アンモニウム水溶液を加え中和した。これをクロロホルム100mlに溶解し、12時間暗所で静置した。得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム:n−ヘキサン=1:2)で精製し、赤褐色液体244mg(収率32%)の目的物を得た。質量スペクトル、ESRにて目的物の生成を確認した。合成を繰り返し、光電変換素子の作製に必要な量を確保した。
【0112】
(2)金属錯体色素を用いた光電変換素子の作製
アセトニトリル(関東化学(株)製、特級)10gに、2−フェニル−2−(4−クロロフェニル)−1,2−ジヒドロキノリン−1−オキシル6.4g、ビス(トリフルオロメタンスルホニル)イミドリチウム(和光純薬工業(株)製)4.4g、ニトロシルテトラフルオロボレート(アルドリッチ社製)225mg、N−(n−ブチル)ベンズイミダゾール(D. Kuang, C. Klein, S. Ito, J.-E. Moser, R. Humphry-Baker, N. Evans, F. Duriaux, C. Gratzel, S. M. Zakeeruddin, M. Gratzel, Adv. Mater. 19, 1133-1137 (2007)に従って合成)1gを加え、電解液(d)を作製した。
【0113】
電解液(a)に代えて電解液(d)を用いた以外は、実施例2と同様にして光電変換素子を作製し評価を行った。得られたI−V曲線を図7に示す。また、I−V曲線の短絡電流(ISC)、開放電圧(VOC)、フィルファクター(FF)及び光電変換効率(η)を表3に示す。
【0114】
【表3】
【0115】
[実施例5]
(1)6−メトキシキノリンから6−メトキシキノリン−1−オキシドへの誘導
6−メトキシキノリン(東京化成工業 (株)製)2.25g(14.13mmol)を200mlのクロロホルムに溶解させた。ここへ、m−クロロ過安息香酸(東京化成工業(株)製、純度65%)2.49g(9.41mmol)を40mlのクロロホルムに溶解させたもの全量を加え、12時間攪拌した。さらに、m−クロロ過安息香酸2.49g(9.41mmol)を40mlのクロロホルムに溶解させたものを加え、12時間攪拌を続けた。その後、1N水酸化ナトリウム水溶液を250ml加え中和した。これに50mlのクロロホルムを加えて抽出した。クロロホルム相を回収し、無水硫酸ナトリウムで乾燥させた。溶媒を除去し、淡黄色固体の目的物2.2g(収率88%)を得た。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0116】
(2)6−メトキシキノリン−1−オキシドから2−フェニル−6−メトキシキノリン−1−オキシドへの誘導
脱水した6−メトキシキノリン−1−オキシド777mg(4.43mmol)を2口ナスフラスコに入れ、窒素置換した。そこへ、脱水テトラヒドロフラン(関東化学(株)製、有機合成用)50mlに溶解させ、これに室温でフェニルマグネシウムブロミド(関東化学(株)製、1.06mol/lテトラヒドロフラン溶液)8.36mlをシリンジで滴下した。反応終了後、飽和塩化アンモニウム水溶液を加えて中和し、クロロホルムにより抽出を行った。3回水洗したのちクロロホルム相を回収し、無水硫酸ナトリウムで乾燥させた。溶媒を除去後、得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 酢酸エチル:ヘキサン=1:2)で精製し淡黄色固体の目的物710mg(収率64%)を得た。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0117】
(3)2−フェニル−6−メトキシキノリン−1−オキシドから2,2−ジフェニル−1,2−ジヒドロキノリン−1−オキシルへの誘導
脱水した2−フェニル−6−メトキシキノリン−1−オキシド300mg(1.19mmol)を2口ナスフラスコに入れ、窒素置換した。そこへ、脱水テトラヒドロフラン(関東化学(株)製、有機合成用)30mlに溶解させ、これに室温でフェニルマグネシウムブロミド(関東化学(株)製、1.06mol/lテトラヒドロフラン溶液)1.68mlをシリンジで滴下した。反応終了後、飽和塩化アンモニウム水溶液を加えて中和し、クロロホルムにより抽出を行った。無水硫酸ナトリウムにて脱水後、エバポレーターにて溶媒を除去した。次に、これをクロロホルム(特級)に溶解させ、24時間室温で攪拌した。その後、中性アルミナカラムクロマトグラフィー(展開溶媒 クロロホルム)で精製し、赤褐色固体の目的物137mg(収率35%)を得た。質量スペクトル、ESRにて目的物の生成を確認した。合成を繰り返し、光電変換素子の作製に必要な量を確保した。
【0118】
(4)金属錯体色素を用いた光電変換素子の作製
FTOガラスLow−E(日本板硝子(株)製)を、3cm×3cmの大きさにダイヤモンドカッターで切り、2−プロパノール(和光純薬工業(株)製、特級)に入れた後、超音波洗浄を5分間行った。超音波洗浄機から取り出して乾燥後、40mM塩化チタン水溶液(和光純薬工業(株)製の塩化チタン(IV)溶液を希釈して使用)に60℃で30分間浸漬した。表面の液体をふき取った後、真空乾燥を1時間行った。ドクターブレード法により、ガラス基板の導電面に厚み15μm設定で二酸化チタンPST−18NR(日揮触媒化成(株)製)を塗布し、5分間室温で放置した。塗布されたガラス基板を125℃のホットプレートに6分間置き、325℃に5分間、375℃に5分間、450℃に15分、500℃に15分間と段階的に温度を上げて焼成した。なお、焼成初期段階で色の変化が認められたが、温度を上げることで透明になった。室温になるまで放置した後、厚み20μm設定で二酸化チタンPST−400C(日揮触媒化成(株)製)をさらに塗布し、5分間室温で放置した。塗布されたガラス基板を125℃のホットプレートに6分間置き、325℃に5分間、375℃に5分間、450℃に15分、500℃に15分間と段階的に温度を上げて焼成した。その後、40mM塩化チタン水溶液(和光純薬工業(株)製の塩化チタン(IV)溶液を希釈して使用)に60℃で30分間浸漬した。表面の液体をふき取った後、真空乾燥を1時間行った。室温になるまで放置した。半導体層を中心部1cm×1cmの範囲を残して削り取った後、再びホットプレートにのせて500℃まで昇温し、30分静置した。その間、シス−ビス(イソチオシアナト)ビス(2,2’−ビピリジル−4,4’−ジカルボキシラト)ルテニウム(II)(アルドリッチ社製)を脱水エタノール(関東化学(株)製、有機合成用)に溶解させ、色素溶液の濃度を0.2mmol/lに調製した。ホットプレートの加熱を止めて80℃になった段階で前記色素溶液中に浸漬して24時間静置し、半導体層に色素を吸着させた。その後、室温にて1時間真空乾燥させて半導体電極を作製した。
【0119】
次に、2箇所に直径1mmの穴をあけたFTO硝子(西野田電工(株)製)を2−プロパノール(和光純薬工業(株)製、特級)に入れた後、超音波洗浄を5分間行った。表面の液体をふき取った後、真空乾燥を1時間行った。ドクターブレード法で、透明ガラス電極作製用白金ペーストPlatisol T/SP(Solaronix社製)をスコッチテープ(3M社製)二枚分に相当する厚みで塗布した。40分間室温で放置し、これをホットプレートに載せ350℃で5分間、450℃で15分間焼成して、対電極を得た。1cm×1cmの穴を開けた熱圧着用封止剤Meltonix 1170−25 Series(Solaronix社製)を用いて半導体電極と対極を140℃のアイロンで接着した。
【0120】
次に、アセトニトリル(関東化学(株)製、特級)に対し、2,2−ジフェニル−6−メトキシ−1,2−ジヒドロキノリン−1−オキシル0.1mol/L、ビス(トリフルオロメタンスルホニル)イミドリチウム(和光純薬工業(株)製)1.2mol/L、ニトロシルテトラフルオロボレート(アルドリッチ社製)0.01mol/L、N−(n−ブチル)ベンズイミダゾール(D. Kuang, C. Klein, S. Ito, J.-E. Moser, R. Humphry-Baker, N. Evans, F. Duriaux, C. Gratzel, S. M. Zakeeruddin, M. Gratzel, Adv. Mater. 19, 1133-1137 (2007)に従って合成)0.5mol/Lとなるようにそれぞれ加え、電解液(e)を作製した。調製した電解液(e)を対極の穴からマイクロシリンジで注入し、穴を接着テープで封じることにより光電変換素子を作製した。
【0121】
この光電変換素子を用いた以外は、実施例1(3)と同様にして評価を行った。得られたI−V曲線を図8に示す。また、I−V曲線の短絡電流(ISC)、開放電圧(VOC)、フィルファクター(FF)及び光電変換効率(η)を、表4に示す。
【0122】
[実施例6]
(1)4−ヨードアニリンからベンジリデン−(4−ヨードアニリン)への誘導
4−ヨードアニリン (東京化成工業(株)製、1級)8.0g(37mmol)を50mlのトルエン(関東化学(株)製、特級)に溶解させた。これに蒸留したベンズアルデヒド(東京化成工業(株)製)3.87g(90.5mmol)を添加し、30分間還流しその後放冷した。n−ヘキサンを加えて析出した粗結晶を濾別し、n−ヘキサンでさらに洗浄した。黄色固体として目的物11.0gを得た(収率98%)。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0123】
(2)ベンジリデン−(4−ヨードアニリン)から2−フェニル−4−エトキシ−6−ヨード−1,2,3,4−テトラヒドロキノリンへの誘導
ベンジリデン−(4−ヨードアニリン)3.0g(9.8mmol)を15mlのトルエン(関東化学(株)製、特級)に溶解させた。蒸留したエチルビニルエーテル(東京化成工業(株)製、1級)1.88mL(19.5mmol)とボロントリフルオリドエタレート(関東化学(株)製)41μL(0.33mmol)を加え二時間還流した。その後、室温に戻して10重量%水酸化ナトリウム水溶液で中和し、クロロホルムを加え、分液の後、有機相を回収した。溶媒を除去後、シリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム)で精製し、黄褐色の液体1.9gを得た(収率51%)。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0124】
(3)2−フェニル−4−エトキシ−6−ヨード−1,2,3,4−テトラヒドロキノリンから2−フェニル−6−ヨードキノリンへの誘導
ナスフラスコに2−フェニル−4−エトキシ−6−ヨード−1,2,3,4−テトラヒドロキノリン1.9g(2.5mmol)とトルエン(関東化学(株)製、特級)10mlを加え溶解させた。室温でジエチルアゾジカルボキシレート(東京化成工業(株)製、40%トルエン溶液)2.2ml(5mmol)を加え12時間還流した。水酸化カリウム水溶液で中和し、クロロホルム分液の後有機相を回収した。無水硫酸ナトリウムによる脱水を行った後、溶媒を除去し、得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム)で精製し、薄黄色の固体1.1g(収率65%)を得た。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0125】
(4)2−フェニル−6−ヨードキノリンから2−フェニル−6−(2,2,2−トリフルオロエトキシ)キノリンへの誘導
2−フェニル−6−ヨードキノリン500mg(1.5mmol)に、トリフルオロエタノール(和光純薬工業(株)製)30mL(420mmol)を加え、さらにヨウ化銅(関東化学(株)製)150mg(0.79mmol)、炭酸セシウム(関東化学(株)製、特級)4.2g(12mmol)、及び2−オキソシクロヘキサカルボン酸エチル(東京化成工業(株)製)250μL(1.57mmol)を加えて14時間還流した。ジクロロメタンと水を加え、分液の後、有機相を回収した。無水硫酸ナトリウムによる脱水を行った後、溶媒を除去し、得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム:n−ヘキサン=5:1)で精製し、白色固体210mg(収率57%)を得た。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0126】
(5)2−フェニル−6−(2,2,2−トリフルオロエトキシ)キノリンから2−フェニル−6−(2,2,2−トリフルオロエトキシ)キノリン−1−オキシドへの誘導
m−クロロ過安息香酸(東京化成工業(株)製、純度65%)90mg(0.3mmol)を2mlのクロロホルムに溶解した溶液を氷冷し、同じく氷冷した2−フェニル−6−(2,2,2−トリフルオロエトキシ)キノリン100mg(0.3mmol)を含む10mlのクロロホルムに滴下し24時間攪拌した。さらに、15時間後にm−クロロ過安息香酸90mg(0.3mmol)を溶解したクロロホルム溶液2mlを加え30時間攪拌した。その後、1N水酸化ナトリウム水溶液を10ml加え中和した。3回水洗したのちクロロホルム相を回収し、無水硫酸ナトリウムで乾燥させた。溶媒を除去後、得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム)で精製し黄白色固体の目的物93mg(収率88%)を得た。質量スペクトル、1H−NMRにて目的物の生成を確認した。
【0127】
(6)2−フェニル−6−(2,2,2−トリフルオロエトキシ)キノリン−1−オキシドから2,2−ジフェニル−6−(2,2,2−トリフルオロエトキシ)−1,2−ジヒドロキノリン−1−オキシルへの誘導
五酸化二リン(関東化学(株)製、1級)で脱水した2−フェニル−6−(2,2,2−トリフルオロエトキシ)キノリン−1−オキシド80mg(0.25mmol)を2口ナスフラスコに入れ、窒素置換した。脱水テトラヒドロフラン(関東化学(株)製、有機合成用)5mlに溶解させ、これに室温でフェニルマグネシウムブロミド(関東化学(株)製、1mol/lテトラヒドロフラン溶液)0.5mlをシリンジで滴下し、1時間攪拌した。反応後、飽和塩化アンモニウム水溶液を加え中和した。これを100mlのクロロホルムに溶解し、12時間暗所で静置した。無水硫酸ナトリウムで乾燥させ、溶媒を除去後、得られた粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム)で精製し、光沢のある赤褐色固体49mg(収率49%)の目的物を得た。質量スペクトル、元素分析、ESRにて目的物の生成を確認した。合成を繰り返し、光電変換素子の作製に必要な量を確保した。
【0128】
次に、アセトニトリル(関東化学(株)製、特級)に対し、2,2−ジフェニル−6−(2,2,2−トリフルオロエトキシ)−1,2−ジヒドロキノリン−1−オキシル0.1mol/L、ビス(トリフルオロメタンスルホニル)イミドリチウム(和光純薬工業(株)製)1.2mol/L、ニトロシルテトラフルオロボレート(アルドリッチ社製)0.01mol/L、N−(n−ブチル)ベンズイミダゾール(D. Kuang, C. Klein, S. Ito, J.-E. Moser, R. Humphry-Baker, N. Evans, F. Duriaux, C. Gratzel, S. M. Zakeeruddin, M. Gratzel, Adv. Mater. 19, 1133-1137 (2007)に従って合成)0.5mol/Lとなるようにそれぞれ加え、電解液(f)を作製した。
電解液(e)に代えて電解液(f)を用いた以外は、実施例5と同様にして光電変換素子を作製し評価を行った。得られたI−V曲線を図9に示す。また、I−V曲線の短絡電流(ISC)、開放電圧(VOC)、フィルファクター(FF)及び光電変換効率(η)を表4に示す。
【0129】
【表4】
【産業上の利用可能性】
【0130】
本発明によって、電解質層の腐食性が低く、かつ、光電変換効率に優れる光電変換素子及びそれを構成するπ共役型有機ラジカル化合物を提供することができる。また、本発明のπ共役型安定有機ラジカル化合物は、様々な有機デバイスへの適用が可能である。例えば有機エレクトロルミネッセンス素子、有機太陽電池、有機トランジスタ、有機メモリー、量子コンピューター等への利用が期待される。
【符号の説明】
【0131】
110 光電変換素子
120 半導体電極
121 半導体層
122 透明導電層
123 電極基材
130 電解質層
140 対電極
150 封止材
【特許請求の範囲】
【請求項1】
半導体層及び色素を含む半導体電極と、対電極と、該半導体電極と該対電極との間に設けられた電解質層とを含む光電変換素子であって、該電解質層が、下記一般式(1)で示されるπ共役型有機ラジカル化合物を含む、上記光電変換素子。
【化1】
(式(1)中、X1、X2、X3、X4、及びX5で表される置換基は、それぞれ独立に電子吸引性基、水素、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、又は炭素数1〜6のフッ素置換アルコキシ基のいずれかである。)
【請求項2】
前記一般式(1)中、X1、X2、X3、X4、及びX5で表される置換基のうち少なくとも1つは電子吸引性基であり、残余の置換基は、それぞれ独立に水素、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、又は炭素数1〜6のフッ素置換アルコキシ基のいずれかである請求項1記載の光電変換素子。
【請求項3】
前記電子吸引性基が、F、Cl、NO2、CN、C6F5、及びCnF2n+1(nは1〜6の整数)からなる群からそれぞれ独立に選ばれた基である請求項2記載の光電変換素子。
【請求項4】
前記色素が金属錯体色素である請求項1〜3のいずれか一項記載の光電変換素子。
【請求項5】
下記一般式(1)で示されるπ共役型有機ラジカル化合物。
【化2】
(式(1)中、X1、X2、X3、X4、及びX5で表される置換基のうち少なくとも1つは電子吸引性基であり、残余の置換基は、それぞれ独立に水素、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、又は炭素数1〜6のフッ素置換アルコキシ基のいずれかである。)
【請求項6】
前記電子吸引性基が、F、Cl、NO2、CN、C6F5、及びCnF2n+1(nは1〜6の整数)からなる群からそれぞれ独立して選ばれた基である請求項5記載のπ共役型有機ラジカル化合物。
【請求項1】
半導体層及び色素を含む半導体電極と、対電極と、該半導体電極と該対電極との間に設けられた電解質層とを含む光電変換素子であって、該電解質層が、下記一般式(1)で示されるπ共役型有機ラジカル化合物を含む、上記光電変換素子。
【化1】
(式(1)中、X1、X2、X3、X4、及びX5で表される置換基は、それぞれ独立に電子吸引性基、水素、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、又は炭素数1〜6のフッ素置換アルコキシ基のいずれかである。)
【請求項2】
前記一般式(1)中、X1、X2、X3、X4、及びX5で表される置換基のうち少なくとも1つは電子吸引性基であり、残余の置換基は、それぞれ独立に水素、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、又は炭素数1〜6のフッ素置換アルコキシ基のいずれかである請求項1記載の光電変換素子。
【請求項3】
前記電子吸引性基が、F、Cl、NO2、CN、C6F5、及びCnF2n+1(nは1〜6の整数)からなる群からそれぞれ独立に選ばれた基である請求項2記載の光電変換素子。
【請求項4】
前記色素が金属錯体色素である請求項1〜3のいずれか一項記載の光電変換素子。
【請求項5】
下記一般式(1)で示されるπ共役型有機ラジカル化合物。
【化2】
(式(1)中、X1、X2、X3、X4、及びX5で表される置換基のうち少なくとも1つは電子吸引性基であり、残余の置換基は、それぞれ独立に水素、炭素数1〜6のアルキル基、炭素数1〜6のアルコキシ基、又は炭素数1〜6のフッ素置換アルコキシ基のいずれかである。)
【請求項6】
前記電子吸引性基が、F、Cl、NO2、CN、C6F5、及びCnF2n+1(nは1〜6の整数)からなる群からそれぞれ独立して選ばれた基である請求項5記載のπ共役型有機ラジカル化合物。
【図1】

【図2】
【図3】

【図4】

【図5】

【図6】

【図7】

【図8】
【図9】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2012−253012(P2012−253012A)
【公開日】平成24年12月20日(2012.12.20)
【国際特許分類】
【出願番号】特願2012−102411(P2012−102411)
【出願日】平成24年4月27日(2012.4.27)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成22年度 独立行政法人科学技術振興機構 研究成果最適展開支援事業 産業技術力強化法第19条の適用を受ける特許出願
【出願人】(000000033)旭化成株式会社 (901)
【出願人】(899000079)学校法人慶應義塾 (742)
【Fターム(参考)】
【公開日】平成24年12月20日(2012.12.20)
【国際特許分類】
【出願日】平成24年4月27日(2012.4.27)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成22年度 独立行政法人科学技術振興機構 研究成果最適展開支援事業 産業技術力強化法第19条の適用を受ける特許出願
【出願人】(000000033)旭化成株式会社 (901)
【出願人】(899000079)学校法人慶應義塾 (742)
【Fターム(参考)】
[ Back to top ]