
新規プロモーター
【課題】環境ストレス、特に生物ストレス及び/又は無生物ストレスに対する誘導性を示す新規なプロモーターを提供する。
【解決手段】(1) 以下の(a)、(b)又は(c)のDNAを含む、環境ストレス応答性プロモーター。(a) 特定の塩基配列からなるDNA(b) 特定の塩基配列において1若しくは複数の塩基が欠失、置換若しくは付加された塩基配列からなり、かつ環境ストレス応答性プロモーターとして機能するDNA(c) 特定の塩基配列と相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ環境ストレス応答性プロモーターとして機能するDNA
【解決手段】(1) 以下の(a)、(b)又は(c)のDNAを含む、環境ストレス応答性プロモーター。(a) 特定の塩基配列からなるDNA(b) 特定の塩基配列において1若しくは複数の塩基が欠失、置換若しくは付加された塩基配列からなり、かつ環境ストレス応答性プロモーターとして機能するDNA(c) 特定の塩基配列と相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ環境ストレス応答性プロモーターとして機能するDNA
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、環境ストレスに対する誘導性を示す新規なプロモーター、当該プロモーターを有する発現ベクター、当該発現ベクターを有する形質転換体並びにトランスジェニック植物、ストレス耐性植物の製造方法、及びスクリーニング方法に関する。
【背景技術】
【0002】
陸生植物は、乾燥、低温、熱、機械的な摂動、創傷及び病原体の感染といった様々な環境ストレスに曝される。これら環境ストレスに植物が曝されると、特定の遺伝子発現といった生化学的、生理学的及び分子的な反応が誘導され、環境ストレスに対する応用性及び順応性を示すことが知られている。このような反応は、植物細胞内における刺激の認識及びシグナル伝達の結果として進行する。しかしながら、このメカニズムの詳細については未だ解明されていないのが現状である。
【発明の開示】
【発明が解決しようとする課題】
【0003】
そこで、本発明は、環境ストレス、特に生物ストレス及び/又は無生物ストレスに対する誘導性を示す新規なプロモーターを提供することを目的とする。
【課題を解決するための手段】
【0004】
本発明者は、上記課題を解決するため鋭意研究を行った結果、cDNAマイクロアレイ分析を応用して、環境ストレスに特異的な発現パターンを示す遺伝子を同定し、当該遺伝子のプロモーター領域を単離することに成功した。また、本発明者は、単離したプロモーターが環境ストレスに応答して、転写制御下の遺伝子に対する誘導性を示すことを見いだし、本発明を完成するに至った。
【0005】
すなわち、本発明は、以下を包含する。
(1) 以下の(a)、(b)又は(c)のDNAを含む、環境ストレス応答性プロモーター。
(a) 配列番号1の塩基配列からなるDNA
(b) 配列番号1の塩基配列において1若しくは複数の塩基が欠失、置換若しくは付加された塩基配列からなり、かつ環境ストレス応答性プロモーターとして機能するDNA
(c) 配列番号1の塩基配列と相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ環境ストレス応答性プロモーターとして機能するDNA
(2) 上記環境ストレスが生物ストレス及び/又は無生物ストレスであることを特徴とする(1)記載のプロモーター。
(3) (1)のプロモーターを含む発現ベクター。
(4) (3)記載の発現ベクターに、さらに任意の遺伝子が組み込まれた発現ベクター。
(5) (3)又は(4)記載の発現ベクターを含む形質転換体。
(6) (3)又は(4)記載の発現ベクターを含むトランスジェニック植物。
(7) 植物が、植物体、植物器官、植物組織又は植物培養細胞である(6)記載のトランスジェニック植物。
(8) (1)のプロモーターと、当該プロモーターによる発現制御可能なかたちで組み込まれたストレス耐性関連遺伝子とを含む発現ベクターでトランスジェニック植物を作製する工程と、
作製したトランスジェニック植物を培養又は栽培する工程とを含む、ストレス耐性植物の製造方法。
(9) (1)記載のプロモーターと、当該プロモーターによる発現制御可能なかたちで組み込まれたレポーター遺伝子とを有する発現ベクターにより形質転換された形質転換体に、検査対象の化合物を接触させる工程と、
上記形質転換体における上記レポーター遺伝子の発現を検出する工程と
を含む、生物ストレス及び/又は無生物ストレスに関与する化合物のスクリーニング方法。
(10) 上記化合物を接触させる工程では、生物ストレス及び/又は無生物ストレスを上記形質転換体に対して負荷することを特徴とする(9)記載のスクリーニング方法。
【発明の効果】
【0006】
本発明によれば、環境ストレス、特に生物ストレス及び/又は無生物ストレスに対する誘導性を示す、全く新規なプロモーターを提供することができる。また、本発明によれば、環境ストレス、特に生物ストレス及び/又は無生物ストレスに対する応答に関与する化合物を同定するための、全く新規なスクリーニング方法を提供することができる。
【発明を実施するための最良の形態】
【0007】
以下、本発明を詳細に説明する。
1.プロモーター
本発明に係るプロモーターは、以下の(a)、(b)又は(c)のDNAを含むものである
(a) 配列番号1の塩基配列からなるDNA
(b) 配列番号1の塩基配列において1若しくは複数の塩基が欠失、置換若しくは付加された塩基配列からなり、かつ環境ストレス応答性プロモーターとして機能するDNA
(c) 配列番号1の塩基配列と相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ環境ストレス応答性プロモーターとして機能するDNA
【0008】
ここで、配列番号1の塩基配列は、シロイヌナズナにおけるRAFL04-16-M08(At3g56710)におけるプロモーター領域を含む塩基配列である。RAFL04-16-M08は、sigA binding proteinをコードする遺伝子である。すなわち、配列番号1の塩基配列は、シロイヌナズナにおけるsigA binding protein遺伝子の開始コドン上流1500塩基からなる。
【0009】
また、上記(b)において、「複数の塩基」とは、1〜10個の塩基、好ましくは1〜5個の塩基を意味する。また、配列番号1の塩基配列において、1若しくは複数の塩基が欠失、置換若しくは付加され得る領域としては、特に限定されないが、配列番号1の塩基配列に含まれる、転写に関与するモチーフ配列を除く領域であることが好ましい。モチーフ配列としては、例えば、G-box、DRE、ABRE、GCC-box、W-box、TGA-box、P-box、MYB及びMYCを挙げることができる。配列番号1の塩基配列におけるこれらのモチーフ配列の存在位置を、図3に示す。図3では各モチーフ配列を下線で示している。
【0010】
さらに上記(c)において、ストリンジェントな条件とは、ナトリウム濃度が25〜500mM、好ましくは25〜300mMであり、温度が42〜68℃、好ましくは42〜65℃である。より具体的には、5×SSC(83mM NaCl、83mMクエン酸ナトリウム)、温度42℃である。
【0011】
また、本発明において、環境ストレスとは、具体的に生物ストレス及び/又は無生物ストレスを意味する。より具体的には、生物ストレスとは、カビ、細菌等が植物に感染することによって当該植物に対して負荷するストレスを意味する。無生物ストレスとは、低温度環境、高温度環境、乾燥環境、高塩濃度環境、高浸透圧環境等に植物を曝すことによって当該植物に対して負荷するストレスを意味する。
【0012】
上記(b)および(c)に示したDNAを含むプロモーターが環境ストレスに対して応答性を示すか否かは、当該プロモーターの下流にGUS遺伝子等のリポーター遺伝子を組み込んだ発現ベクターを用いて形質転換体を作製し、当該形質転換体が環境ストレスに曝された状態で上記リポーター遺伝子を発現しているか否かを検討することによって確認することができる。
【0013】
具体的に環境ストレスとしては、サリチル酸で処理する方法、プロベナゾールで処理する方法、バイオン、パラコート、重金属で処理する方法を挙げることができる。また、リポーター遺伝子としてGUS遺伝子を使用した場合には、環境ストレスに曝した後の形質転換植物におけるGUS活性を測定する。このような条件で環境ストレスに曝すことで、供試プロモーターが環境ストレスに対して応答性を示すか否かを判定することができる。
【0014】
2.発現ベクター
本発明の発現ベクターは、適当なベクターに本発明のプロモーターを連結(挿入)することにより得ることができる。本発明のプロモーターを挿入するためのベクターは、宿主中で複製可能なものであれば特に限定されず、例えばプラスミド、シャトルベクター、ヘルパープラスミドなどが挙げられる。
【0015】
プラスミドDNAとしては、大腸菌由来のプラスミド(例えばpBR322、pBR325、pUC118、pUC119、pUC18、pUC19、pBluescript等)、枯草菌由来のプラスミド(例えばpUB110、pTP5等)、酵母由来のプラスミド(例えばYEp13、YCp50等)などが挙げられ、ファージDNAとしてはλファージ(Charon4A、Charon21A、EMBL3、EMBL4、λgt10、λgt11、λZAP等)が挙げられる。さらに、レトロウイルス又はワクシニアウイルスなどの動物ウイルス、バキュロウイルスなどの昆虫ウイルスベクターを用いることもできる。
【0016】
ベクターに本発明のプロモーターを挿入するには、まず、精製されたDNAを適当な制限酵素で切断し、適当なベクター DNAの制限酵素部位又はマルチクローニングサイトに挿入してベクターに連結する方法などが採用される。
【0017】
本発明においては、任意遺伝子を発現させるため、上記発現ベクターに、さらに当該任意遺伝子を挿入することができる。任意の遺伝子を挿入する手法は、ベクターにプロモーターを挿入する方法と同様である。
【0018】
本発明のプロモーターは、その3'末端にレポーター遺伝子、例えば、植物で広く用いられているGUS遺伝子を連結して用いれば、GUS活性を調べることでプロモーターの強さを容易に評価することができる。なお、レポーター遺伝子としては、GUS遺伝子以外にも、ルシフェラーゼ、グリーンフルオレセイントプロテインなども用いることができる。
【0019】
このように、本発明においては、様々なベクターを用いることができる。さらに、本発明のプロモーターに目的の任意遺伝子をセンス又はアンチセンス方向で接続したものを作製し、これをバイナリーベクターと呼ばれるpBI101(Clonetech社)などのベクターに挿入することができる。
【0020】
3.形質転換体の作製
本発明の形質転換体は、本発明の発現ベクターを宿主中に導入することにより得ることができる。ここで、宿主としては、上述したプロモーターが機能しうるものであれば特に限定されるものではないが、植物が好ましい。宿主が植物である場合は、形質転換植物(トランスジェニック植物)は以下のようにして得ることができる。
【0021】
本発明において形質転換の対象となる植物は、植物体全体、植物器官(例えば葉、花弁、茎、根、種子等)、植物組織(例えば表皮、師部、柔組織、木部、維管束等)又は植物培養細胞のいずれをも意味するものである。形質転換に用いられる植物としては、アブラナ科、イネ科、ナス科、マメ科等に属する植物(下記参照)が挙げられるが、これらの植物に限定されるものではない。
アブラナ科:シロイヌナズナ(Arabidopsis thaliana)
ナス科:タバコ(Nicotiana tabacum)
イネ科:トウモロコシ(Zea mays) 、イネ(Oryza sativa)
マメ科:ダイズ(Glycine max)
【0022】
上記発現ベクターは、通常の形質転換方法、例えば電気穿孔法(エレクトロポレーション法)、アグロバクテリウム法、パーティクルガン法、PEG法等によって植物中に導入することができる。
【0023】
例えばエレクトロポレーション法を用いる場合は、パルスコントローラーを備えたエレクトロポレーション装置により、電圧500〜1600V、25〜1000μF、20〜30msecの条件で処理し、遺伝子を宿主に導入する。
【0024】
また、パーティクルガン法を用いる場合は、植物体、植物器官、植物組織自体をそのまま使用してもよく、切片を調製した後に使用してもよく、プロトプラストを調製して使用してもよい。このように調製した試料を遺伝子導入装置(例えばBio-Rad社のPDS-1000/He等)を用いて処理することができる。処理条件は植物又は試料により異なるが、通常は1000〜1800psi程度の圧力、5〜6cm程度の距離で行う。
【0025】
また、植物ウイルスをベクターとして利用することによって、本発明に係るプロモーターの発現制御下に目的遺伝子を植物体に導入することができる。利用可能な植物ウイルスとしては、例えば、カリフラワーモザイクウイルスが挙げられる。すなわち、まず、ウイルスゲノムを大腸菌由来のベクターなどに挿入して組換え体を調製した後、ウイルスのゲノム中に、これらの目的遺伝子を挿入する。このようにして修飾されたウイルスゲノムを制限酵素によって組換え体から切り出し、植物宿主に接種することによって、目的遺伝子を植物宿主に導入することができる。
【0026】
アグロバクテリウムのTiプラスミドを利用する方法においては、アグロバクテリウム(Agrobacterium)属に属する細菌が植物に感染すると、それが有するプラスミドDNAの一部を植物ゲノム中に移行させるという性質を利用して、本発明に係るプロモーター及び目的遺伝子を植物宿主に導入する。アグロバクテリウム属に属する細菌のうちアグロバクテリウム・ツメファシエンス(Agrobacterium tumefaciens)は、植物に感染してクラウンゴールと呼ばれる腫瘍を形成し、また、アグロバクテリウム・リゾゲネス(Agrobacteriumu rhizogenes)は、植物に感染して毛状根を発生させる。これらは、感染の際にTiプラスミド又はRiプラスミドと呼ばれる各々の細菌中に存在するプラスミド上のT-DNA領域(Transferred DNA)と呼ばれる領域が植物中に移行し、植物のゲノム中に組み込まれることに起因するものである。
【0027】
Ti又はRiプラスミド上のT-DNA領域中に、植物ゲノム中に組み込みたいDNAを挿入しておけば、アグロバクテリウム属の細菌が植物宿主に感染する際に目的とするDNAを植物ゲノム中に組込むことができる。
【0028】
形質転換の結果得られる腫瘍組織やシュート、毛状根などは、そのまま細胞培養、組織培養又は器官培養に用いることが可能であり、また従来知られている植物組織培養法を用い、適当な濃度の植物ホルモン(オーキシン、サイトカイニン、ジベレリン、アブシジン酸、エチレン、ブラシノライド等)の投与などにより植物体に再生させることができる。
【0029】
ここで、本発明に係るプロモーターによる発現制御下で導入される目的遺伝子としては、特に限定されないが、例えば、植物のストレス耐性に関連する遺伝子(ストレス耐性関連遺伝子と呼ぶ)を挙げることができる。ストレス耐性関連遺伝子としては、例えば、抗菌性タンパク質をコードするキチナーゼ遺伝子、β-1,3-グルカナーゼ遺伝子、さらには、ストレス耐性関連遺伝子の発現を制御する転写因子WRKY、ERF等を挙げることができる。これらのなかから選ばれる少なくとも1種のストレス耐性関連遺伝子を、本発明に係るプロモーターにより発現制御可能となるように宿主に導入することによって、ストレス耐性形質転換体を作製することができる。宿主として植物を使用した場合には、ストレス耐性植物を製造することができる。
【0030】
ところで、本発明に係るプロモーターと、当該プロモーターによる発現制御可能なかたちで組み込まれたレポーター遺伝子とを有する発現ベクターにより形質転換された形質転換体に検査対象の化合物を接触させ、形質転換体におけるレポーター遺伝子の発現を検出することによって、生物ストレス及び/又は無生物ストレスに関与する化合物をスクリーニングすることができる。ここで、検査対象の化合物を形質転換体に接触させる際には、生物ストレス及び/又は無生物ストレスを負荷した状態であってもよい。
【0031】
本発明の発現ベクターは、上記植物宿主に導入するのみならず、大腸菌(Escherichia coli)等のエッシェリヒア属、バチルス・ズブチリス(Bacillus subtilis)等のバチルス属、又はシュードモナス・プチダ(Pseudomonas putida)等のシュードモナス属に属する細菌、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)、シゾサッカロミセス・ポンベ(Schizosaccharomyces pombe)等の酵母、COS細胞、CHO細胞等の動物細胞、あるいはSf9等の昆虫細胞などに導入して形質転換体を得ることもできる。大腸菌、酵母等の細菌を宿主とする場合は、本発明の発現ベクターが該細菌中で自律複製可能であると同時に、本発明のプロモーター、リボソーム結合配列、目的遺伝子、転写終結配列により構成されていることが好ましい。また、プロモーターを制御する遺伝子が含まれていてもよい。
【0032】
細菌への組換えベクターの導入方法は、細菌にDNAを導入する方法であれば特に限定されるものではない。例えばカルシウムイオンを用いる方法、エレクトロポレーション法等が挙げられる。
【0033】
酵母を宿主とする場合は、例えばサッカロミセス・セレビシエ(Saccharomyces cerevisiae)、シゾサッカロミセス・ポンベ(Schizosaccharomyces pombe)などが用いられる。酵母への組換えベクターの導入方法は、酵母にDNAを導入する方法であれば特に限定されず、例えばエレクトロポレーション法、スフェロプラスト法、酢酸リチウム法等が挙げられる。
【0034】
動物細胞を宿主とする場合は、サル細胞COS-7、Vero、チャイニーズハムスター卵巣細胞(CHO細胞)、マウスL細胞などが用いられる。動物細胞への組換えベクターの導入方法としては、例えばエレクトロポレーション法、リン酸カルシウム法、リポフェクション法等が挙げられる。
【0035】
昆虫細胞を宿主とする場合は、Sf9細胞などが用いられる。昆虫細胞への組換えベクターの導入方法としては、例えばリン酸カルシウム法、リポフェクション法、エレクトロポレーション法などが挙げられる。
【0036】
遺伝子が宿主に組み込まれたか否かの確認は、PCR法、サザンハイブリダイゼーション法、ノーザンハイブリダイゼーション法等により行うことができる。例えば、形質転換体からDNAを調製し、DNA特異的プライマーを設計してPCRを行う。PCRは、前記プラスミドを調製するために使用した条件と同様の条件で行われる。その後は、増幅産物についてアガロースゲル電気泳動、ポリアクリルアミドゲル電気泳動又はキャピラリー電気泳動等を行い、臭化エチジウム、SYBR Green液等により染色し、そして増幅産物を1本のバンドとして検出することにより、形質転換されたことを確認する。また、予め蛍光色素等により標識したプライマーを用いてPCRを行い、増幅産物を検出することもできる。さらに、マイクロプレート等の固相に増幅産物を結合させ、蛍光又は酵素反応等により増幅産物を確認する方法も採用してもよい。
【0037】
4.植物の製造
本発明においては、上記形質転換植物細胞等から形質転換植物体に再生することができる。再生方法としては、カルス状の形質転換細胞をホルモンの種類、濃度を変えた培地へ移して培養し、不定胚を形成させ、完全な植物体を得る方法が採用される。使用する培地としては、LS培地、MS培地などが例示される。
【0038】
本発明の「植物体を製造する方法」は、上記植物プロモーターを挿入した植物発現ベクターを宿主細胞に導入して形質転換植物細胞を得て、該形質転換植物細胞から形質転換植物体を再生し、得られた形質転換植物体から植物種子を得て、該植物種子から植物体を生産する工程を含む。
【0039】
形質転換植物体から植物種子を得るには、例えば、形質転換植物体を発根培地から採取し、水を含んだ土を入れたポットに移植し、一定温度下で生育させて、花を形成させ、最終的に種子を形成させる。また、種子から植物体を生産するには、例えば、形質転換植物体上で形成された種子が成熟したところで、単離して、水を含んだ土に播種し、一定温度、照度下で生育させることにより、植物体を生産する。このようにして育種された植物は、導入されたプロモーターのストレス応答性に応じた環境ストレス耐性植物となる。
【実施例】
【0040】
以下、実施例を用いて本発明をより詳細に説明するが、本発明の技術的範囲は以下の実施例に限定されるものではない。
【0041】
〔実施例1〕マイクロアレイによる発現解析
本発明者らは、シロイヌナズナの約7,000個の完全長cDNAを有するマイクロアレイ(関ら、Plant J. 31, 279-292 (2002)に開示されたマイクロアレイ)を用いて、生物ストレス或いは無生物ストレスを負荷した条件下で特異的に発現している遺伝子を網羅的に解析した。本例で使用したマイクロアレイは、Arabidopsisの完全長cDNAライブラリーから単離した遺伝子に加えて、RD(Responsive to Dehydration)遺伝子、ERD(Early Responsive to Dehydration)遺伝子、内部標準としてλコントロール鋳型DNA断片(TX803、宝酒造株式会社製)、さらにネガティブコントロールとしてマウスのニコチン酸アセチルコリンレセプターのエプシロンサブユニット(nAChRE)遺伝子及びマウスのグルココルチコイドレセプターの相同性遺伝子からなる計約7000のcDNAを有している。
【0042】
このマイクロアレイを用いて、以下のように環境ストレスに対して応答性を示す遺伝子を解析した。実験に用いたシロイヌナズナは、2%Murashige and Skoog塩(和光純薬社製)、2%スクロース及び0.8%バクトアガー(Difco社製)を含むGM培地で発芽させ、その後、16時間(午前5時〜午後9時)を明下とし8時間を暗下とするサイクルで、22℃のチャンバー内で3週間成長させた。また、様々な環境ストレス処理は、ほぼ同時刻(午前9時〜午前10時の間)に開始した。
【0043】
本実験において、〔ストレス負荷時の発現量〕/〔ストレスがない時の発現量〕の値がλコントロール鋳型DNA断片と比較して3倍以上であり、且つ、少なくとも一回の実験で1000以上のシグナル値を示すものを環境ストレス負荷によって特異的に発現する遺伝子と認定した。本例においては、生物ストレス負荷時及び無生物ストレス負荷時において特異的に発現する遺伝子を解析した。
【0044】
生物ストレスとしては、非病原性糸状菌(Alternaria alternata O-276;鳥取大学の尾谷氏より供与)、不親和性糸状菌(Alternaria brassicicola O-264;鳥取大学の尾谷氏より供与)及び親和性糸状菌(Colletotrichum higginsianum MAFF 305635;MAFF Genebankより入手)を接種することによって行った。無生物ストレスは、乾燥処理、低温処理、高塩濃度処理、UV-C処理及びCuSO4処理によって行った。これら無生物ストレス及び生物ストレスは、“The cDNA Microarray Analysis Using an Arabidopsis pad3 Mutant Reveals the Expression Profiles and Classification of Genes Induced by Alternaria brassicicola Attack”(鳴坂らPlant Cell Physiol. 44(4) 377-387 (2003))に記載の方法に準じて行った。
【0045】
シロイヌナズナからのtotal RNAの抽出は、生物ストレス負荷から10、24及び48時間後に行い、無生物ストレス負荷から2、5、10及び24時間後に行った。また、コントロールとして、蒸留水処理から2、5、10、24及び48時間後の植物からもtotal RNAを抽出した。なお、total RNAの抽出にはTRIZOL Reagent(Invitrogen社製)を使用し、total RNAからのmRNAの抽出にはmRNA isolation kit(Miltenyi Biotec社製)を使用した。
【0046】
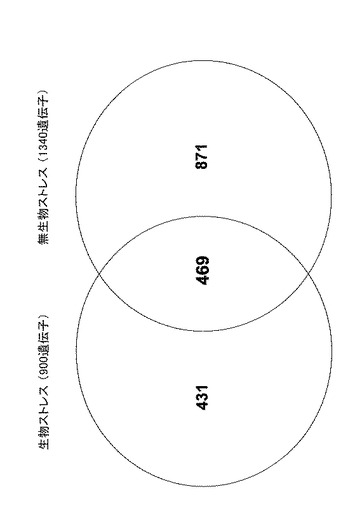
その結果、生物ストレス負荷によって900個の遺伝子が特異的に発現していることが判った。なお、Alternaria alternataの接種によって118個の遺伝子が特異的に発現し、Alternaria brassicicolaの接種によって864個の遺伝子が特異的に発現し、Colletotrichum higginsianumの接種によって142個の遺伝子が特異的に発現した。これに対して、無生物ストレス負荷によって1340個の遺伝子が特異的に発現していることが判った。なお、乾燥処理によって740個の遺伝子が特異的に発現し、低温処理によって438個の遺伝子が特異的に発現し、高塩濃度処理によって605個の遺伝子が特異的に発現し、UV-C処理によって290個の遺伝子が特異的に発現し、CuSO4処理によって676個の遺伝子が特異的に発現した。これらの結果を図1にまとめる。図1から判るように、生物ストレス及び無生物ストレスによって特異的に発現する遺伝子の数は469個であった。
【0047】
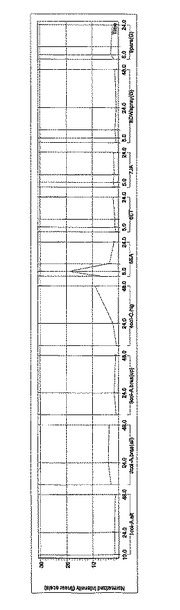
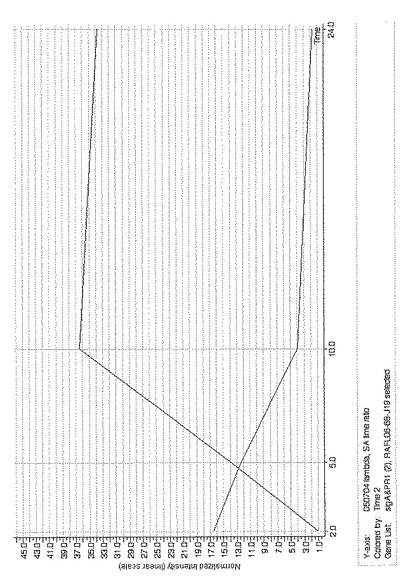
この469個の遺伝子のうち、RAFL04-16-M08については、サリチル酸処理特異的に発現していることが明らかとなった。生物ストレス及び無生物ストレスによるRAFL04-16-M08の発現量の変化を図2に示す。なお、図2において縦軸は規格化した強度、すなわち、発現量の増加を示し、横軸はストレス負荷からの時間を示している。
【0048】
なお、RAFL04-16-M08は、SigA binding protein(At3g56710)である。以下、RAFL04-16-M08を「SigA BP遺伝子」と称する。なお、実施例中、低温処理は、4℃で生育させる条件で行った。高塩濃度処理はシロイヌナズナを250mMのNaCl溶液で水耕栽培する条件で行った。UV-C処理はシロイヌナズナに1kJの紫外線を照射する条件で行った。CuSO4処理は10mMのCuSO4溶液を噴霧する条件で行った。アブシジン酸処理は100μMのABA溶液で水耕栽培する条件とした。サリチル酸処理は5mMのサリチル酸溶液を噴霧する条件で行った。ジャスモン酸処理は200μMのジャスモン酸メチル溶液で水耕栽培する条件とした。エチレン処理(エテフォン処理)は1mMのエテフォン溶液で水耕栽培する条件とした。パラコート処理は25μMのパラコート溶液を噴霧する条件で行った。ローズベンガル処理は20mMのローズベンガル溶液を噴霧する条件で行った。
【0049】
以上、本例によってSigA BP遺伝子は生物ストレス及び無生物ストレスのいずれを負荷しても発現が誘導されることが明らかとなり、当該SigA BP遺伝子の上流に存在するプロモーターが生物ストレス及び無生物ストレス誘導性のプロモーターであることが推測された。
【0050】
〔実施例2〕マイクロアレイによる発現解析2
本発明者らは、シロイヌナズナの約7,000個の完全長cDNAを有するマイクロアレイ(関ら、Plant J. 31, 279-292 (2002)に開示されたマイクロアレイ)を用いて、生物ストレス或いは無生物ストレスを負荷した条件下で特異的に発現している遺伝子を網羅的に解析した。本例で使用したマイクロアレイは、Arabidopsisの完全長cDNAライブラリーから単離した遺伝子に加えて、RD(Responsive to Dehydration)遺伝子、ERD(Early Responsive to Dehydration)遺伝子、内部標準としてλコントロール鋳型DNA断片(TX803、宝酒造株式会社製)、さらにネガティブコントロールとしてマウスのニコチン酸アセチルコリンレセプターのエプシロンサブユニット(nAChRE)遺伝子及びマウスのグルココルチコイドレセプターの相同性遺伝子からなる計約7000のcDNAを有している。
【0051】
このマイクロアレイを用いて、以下のように環境ストレスに対して応答性を示す遺伝子を解析した。植物材料として、土(ダイオ化成社製)に播種し、24時間の明暗サイクルを明時間16時間及び暗時間8時間として4週間栽培したシロイヌナズナ(生態型:コロンビア)を用いた。また、様々な環境ストレス処理は、ほぼ同時刻(午前9時〜午前10時の間)に開始した。
【0052】
本実験において、〔ストレス負荷時の発現量〕/〔ストレスがない時の発現量〕の値がλコントロール鋳型DNA断片と比較して3倍以上であり、且つ、少なくとも一回の実験で1000以上のシグナル値を示すものを環境ストレス負荷によって特異的に発現する遺伝子と認定した。本例においては、生物ストレス負荷時及び無生物ストレス負荷時において特異的に発現する遺伝子を解析した。
【0053】
生物ストレスとしては、非病原性糸状菌(Alternaria alternata O-276;鳥取大学の尾谷氏より供与)、不親和性糸状菌(Alternaria brassicicola O-264;鳥取大学の尾谷氏より供与)及び親和性糸状菌(Colletotrichum higginsianum MAFF 305635;MAFF Genebankより入手)を接種することによって行った。無生物ストレスは、乾燥処理、低温処理、高塩濃度処理、UV-C処理及びCuSO4処理によって行った。これら無生物ストレス及び生物ストレスは、“The cDNA Microarray Analysis Using an Arabidopsis pad3 Mutant Reveals the Expression Profiles and Classification of Genes Induced by Alternaria brassicicola Attack”(鳴坂らPlant Cell Physiol. 44(4) 377-387 (2003))に記載の方法に準じて行った。
【0054】
シロイヌナズナからのtotal RNAの抽出は、生物ストレス負荷から10、24及び48時間後に行い、無生物ストレス負荷から2、5、10及び24時間後に行った。また、コントロールとして、蒸留水処理から2、5、10、24及び48時間後の植物からもtotal RNAを抽出した。なお、total RNAの抽出にはTRIZOL Reagent(Invitrogen社製)を使用し、total RNAからのmRNAの抽出にはmRNA isolation kit(Miltenyi Biotec社製)を使用した。
【0055】
発現遺伝子のうち、RAFL04-16-M08については、サリチル酸処理特異的に発現していることが明らかとなった。生物ストレス及び無生物ストレスによるRAFL04-16-M08の発現量の変化を図2に示す。なお、図2において縦軸は規格化した強度、すなわち、発現量の増加を示し、横軸はストレス負荷からの時間を示している。
【0056】
サリチル酸処理は5mMのサリチル酸溶液を噴霧する条件で行った。ジャスモン酸処理は100μMのジャスモン酸メチル溶液を噴霧する条件で行った。エチレン処理(エテフォン処理)は1mMのエテフォン溶液を噴霧する条件で行った。パラコート処理は25μMのパラコート溶液を噴霧する条件で行った。
【0057】
以上、本例によってSigA BP遺伝子は生物ストレス及び無生物ストレスのいずれを負荷しても発現が誘導されることが明らかとなり、当該SigA BP遺伝子の上流に存在するプロモーターが生物ストレス及び無生物ストレス誘導性のプロモーターであることが推測された。
【0058】
〔実施例3〕SigA BP遺伝子のプロモーター領域の解析
本例では、生物ストレス及び無生物ストレス誘導性のプロモーターであると推測された、SigA BP遺伝子の上流に存在するプロモーターを単離し、そのプロモーター活性を解析した。
【0059】
先ず、SigA BP遺伝子のcDNAの塩基配列を定法に従って決定した。次に、この塩基配列情報に基づいてシロイヌナズナゲノム・データベース(URL; http://rarge.gsc.riken.go.jp/)を検索し、SigA BP遺伝子の開始コドンより上流約1.5kbをプロモーターとして同定した。シロイヌナズナにおいては、開始コドンより上流約1.0kb内にプロモーターが存在し、開始コドンより約150b内に5’非翻訳領域が存在することが知られているためである(MaleckらNature Genet. 26, 403-410 (2000))。
【0060】
本例で同定したプロモーター領域の塩基配列を配列番号1及び図3に示す。なお、図3には、塩基配列情報に基づいて特定されたシスエレメント領域を、下線で示している。また、下線の下に付記した文字はシスエレメントの種類を示している。図3中「W-box」はWRKY転写因子の結合領域を示している。WRKY転写因子は、病害防御、老化及びトライコーム形成等の様々な生理学的問題の制御に関わっており(EulgemらTrends Plant Sci. 5, 199-206 (2000))、生物ストレスに対する応答性に寄与するものと考えられた。図3中「ACGT」及び「TGA-box」はactivation sequence-1 (as-1)因子のコア配列(TGACGT/C)を示している(Lam et al.,(1989)Proc. Natl. Acad. Sci. USA 86, 7890-7894)。なおas-1因子は酸化ストレス応答性因子として作用する。「ACGT」及び「TGA-box」は、活性酸素種応答性プロモーター内においてシスエレメントとして作用すると考えられる(GarretonらPlant Physiol. 130, 1516-1526 (2002))。図3中の「G-box」はサリチル酸(Maleck, K. et al (2000) Nature Genet. 26, 403-410.)、アブシジン酸’Guiltinan, M.J. et al. (1990) Science 250, 267-271.)、エチレン(Eyal, Y. et al. (1993) Plant J. 4, 225-234.)及びジャスモン酸(Mason, H.S. et al. (1993) Plant Cell 5, 241-251.)に応答するシスエレメント(ACGTG)として知られている。図3中の「P-box」は病害防御応答に関するシスエレメント(CCA/TA/TCC)して作用する。
【0061】
次に、配列番号1に示した塩基配列からなるプロモーターが無生物ストレス及び/又は生物ストレスに対して誘導性を示すか否かを検討した。配列番号1に示した塩基配列からなるプロモーター及び当該プロモーターの下流に位置するGUS遺伝子を有するベクターを以下のように作製した。
【0062】
シロイヌナズナゲノムDNAからPCRを用いて、SigA BP遺伝子の開始コドンより上流1.5kbをSigA BP遺伝子のプロモーターとしてクローニングした。PCRはプライマー配列に制限酵素Hind IIIの認識部位を付与したプライマー(プライマー1:cccaagcttccattcgtttcttaatccttacc(配列番号2))とBamHIの認識部位を付与したプライマー(プライマー2:cgggatccgttcggttcctgtaagagaattcag(配列番号3))を使用して、最初に94℃で2分反応させた後、94℃で30秒、55℃で30秒及び74℃で2分のサイクルを25サイクル、最後に74℃で7分の反応条件で行った。
【0063】
このSigA BP遺伝子のプロモーターDNAを制限酵素Hind IIIとBamHIで切断し、バイナリーベクターと呼ばれるpBI101(Clonetech社)ベクター DNAのマルチクローニングサイト、制限酵素Hind IIIとBamHI切断部位に挿入してベクターに連結した。これによりSigA BP遺伝子のプロモーターの3'末端にレポーター遺伝子、GUS遺伝子を連結した、ベクターを構築した。
【0064】
〔実施例4〕プロモーター活性の検定
このように作製したベクターを用いてアグロバクテリウムを介してシロイヌナズナを形質転換し、最終的に形質転換シロイヌナズナホモ接合体株を得た。得られたホモ接合体株を播種後25〜28日経過した時点で、マイクロアレイ解析によってSigA BP遺伝子を特異的に誘導することがあきらかとなったサリチル酸で処理した後、GUS活性を測定することによって、プロモーター活性を評価した。具体的には、0.5mMのサリチル酸またはコントロールとして蒸留水10mlをシロイヌナズナ形質転換体に散布した。散布24時間後にロゼット葉1〜2枚程度を1.5ml チューブに入れ、200μlの抽出用緩衝液(50mM リン酸ナトリウム緩衝液pH7.0, 10mM EDTA, 0.1%Triton X-100, 10mM 2-メルカプトエタノール)を添加し、氷上でホモジナイザーにて破砕した。
【0065】
15000rpm, 4℃,で10分間遠心した後、上清10μlを2mlチューブに移し、基質として100μlの4-methylumbelliferyl β-D-glucuronide (4-MUG)(0.4mg/ml抽出用緩衝液)を加え、37℃で1時間反応した。1時間後、1.4mlの0.2M Na2CO3を添加して反応を停止した。その後、365nmの励起光による455nmの放出スペクトルを測定することにより、単位時間、単位タンパク質含量当たりの4-MUの生成量(pmol 4-MU/min/mg protein)を求めた。上清中のタンパク質含量はBioRad protein assay Kitを使用して、BSAをスタンダードとして、添付のマニュアルにしたがって定量した。
【0066】
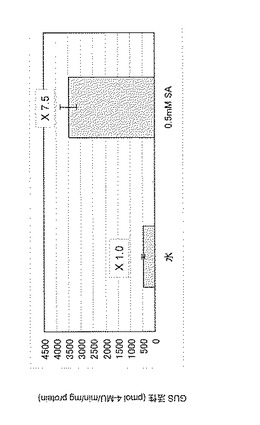
その結果、図4に示すように、蒸留水噴霧処理と比較して、サリチル酸処理によってGUS活性は増強していることが判った。なお図4において、活性値は3サンプルの平均±標準偏差で示しており、各棒グラフ上の数値は水処理でのGUS活性を1.0としたときの各薬剤処理でのGUS活性の相対比を表している。
【0067】
〔実施例5〕PR-1プロモーターとの比較検定-1
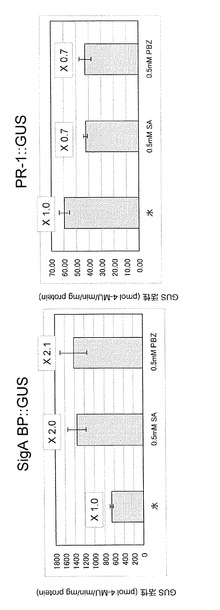
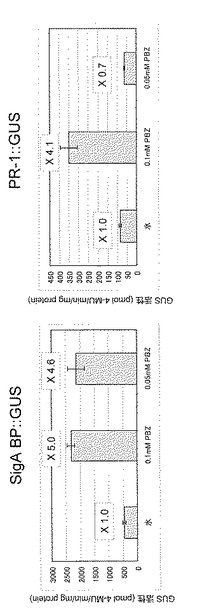
0.5mMのサリチル酸、0.5mMのプロベナゾールまたはコントロールとして蒸留水10mlを 実施例4と同様に栽培したSigA BP::GUSおよびPR-1::GUS形質転換シロイヌナズナ(いずれもホモ接合体株)の株元に滴下した。処理2時間後に実施例4で示した方法に従ってGUS活性を測定した。その結果、図5に示すように、SigA BP::GUS形質転換シロイヌナズナでは蒸留水噴霧処理と比較して、サリチル酸およびプロベナゾール処理によってGUS活性は増強していることが判った。一方、PR-1::GUS形質転換シロイヌナズナではGUS活性の増強は認められなかった。新規なplant activatorを見出すには多数の試験化合物を効率よく検定することが重要である。本実施例で示されたように、SigA BPの発現を指標とすることによって、従来のマーカー遺伝子(PR-1)に比較して短時間で化合物の検定を行うことが可能となる。なお図5において、活性値は3サンプルの平均±標準偏差で示しており、各棒グラフ上の数値は水処理でのGUS活性を1.0としたときの各薬剤処理でのGUS活性の相対比を表している。
【0068】
本実施例の結果は図6に示したサリチル酸処理後のSigA BP およびPR-1遺伝子のマイクロアレイ解析による発現の経時変化からも支持される。すなわち、SigA BPはサリチル酸処理2時間後には遺伝子発現が認められているのに対して、PR-1は有意な遺伝子発現が認められていない。
【0069】
なお、PR-1::GUS形質転換シロイヌナズナは以下の方法で構築したベクターを用いてアグロバクテリウムを介して作出した。
【0070】
シロイヌナズナゲノムDNAからPCRを用いて、PR-1遺伝子の開始コドンより上流1290bpをPR-1遺伝子のプロモーターとしてクローニングした。PCRはプライマー配列に制限酵素Hind IIIの認識部位を付与したプライマー(プライマー1:gcacaagcttgttttaac(配列番号4))とXbaIの認識部位を付与したプライマー(プライマー2:gagatctagattttctaagttgataatggt(配列番号5))を使用して、最初に94℃で2分反応させた後、94℃で30秒、55℃で30秒及び74℃で1分のサイクルを30サイクル、最後に74℃で7分の反応条件で行った。
【0071】
このPR-1遺伝子のプロモーターDNAを制限酵素Hind IIIとXbaIで切断し、バイナリーベクターと呼ばれるpBI101(Clonetech社)ベクター DNAのマルチクローニングサイト、制限酵素Hind IIIとXbaI切断部位に挿入してベクターに連結した。これによりPR-1遺伝子のプロモーターの3'末端にレポーター遺伝子、GUS遺伝子を連結した、ベクターを構築した。
【0072】
〔実施例6〕PR-1プロモーターとの比較検定-2
0.1mMおよび0.05 mM のプロベナゾールまたはコントロールとして蒸留水10mlを実施例4と同様に栽培したSigA BP::GUS形質転換シロイヌナズナ(ホモ接合体株)の株元に滴下した。処理1日後に実施例4で示した方法に従ってGUS活性を測定した。その結果、図7に示すように、SigA BP::GUS形質転換シロイヌナズナでは蒸留水噴霧処理と比較して、0.05mM プロベナゾール処理でもGUS活性は増強していることが判った。一方、PR-1::GUS形質転換シロイヌナズナでは0.1 mM 処理ではGUS活性の増強が認められたが、0.05mM処理では認められなかった。本実施例で示されたように、SigA BPの発現を指標とすることによって従来のマーカー遺伝子(PR-1)では評価できないポテンシャルの低いplant activatorのリード化合物を見出すことが可能となる。なお、図7において活性値は3サンプルの平均±標準偏差で示しており、各棒グラフ上の数値は水処理でのGUS活性を1.0としたときの各薬剤処理でのGUS活性の相対比を表している。
【図面の簡単な説明】
【0073】
【図1】生物ストレス誘導性の遺伝子及び無生物ストレス誘導性の遺伝子をマイクロアレイで解析した結果を分類したベン図である。
【図2】生物ストレス誘導性及び無生物ストレスによるSigA BP遺伝子の発現量の変化を示す特性図である。
【図3】本発明に係るプロモーターの塩基配列を解析した結果を示す図である。
【図4】本発明に係るプロモーターの活性を、GUS遺伝子の発現に起因するGUS活性で測定した結果を示す特性図である。
【図5】本発明に係るプロモーターの活性を、PR-1プロモーターと比較した結果を示す特性図である。
【図6】サリチル酸散布によるSigA BPおよびPR-1遺伝子の発現量の変化を示す特性図である。
【図7】本発明に係るプロモーターの薬剤濃度に対する感受性を、PR-1プロモーターと比較した結果を示す特性図である。
【技術分野】
【0001】
本発明は、環境ストレスに対する誘導性を示す新規なプロモーター、当該プロモーターを有する発現ベクター、当該発現ベクターを有する形質転換体並びにトランスジェニック植物、ストレス耐性植物の製造方法、及びスクリーニング方法に関する。
【背景技術】
【0002】
陸生植物は、乾燥、低温、熱、機械的な摂動、創傷及び病原体の感染といった様々な環境ストレスに曝される。これら環境ストレスに植物が曝されると、特定の遺伝子発現といった生化学的、生理学的及び分子的な反応が誘導され、環境ストレスに対する応用性及び順応性を示すことが知られている。このような反応は、植物細胞内における刺激の認識及びシグナル伝達の結果として進行する。しかしながら、このメカニズムの詳細については未だ解明されていないのが現状である。
【発明の開示】
【発明が解決しようとする課題】
【0003】
そこで、本発明は、環境ストレス、特に生物ストレス及び/又は無生物ストレスに対する誘導性を示す新規なプロモーターを提供することを目的とする。
【課題を解決するための手段】
【0004】
本発明者は、上記課題を解決するため鋭意研究を行った結果、cDNAマイクロアレイ分析を応用して、環境ストレスに特異的な発現パターンを示す遺伝子を同定し、当該遺伝子のプロモーター領域を単離することに成功した。また、本発明者は、単離したプロモーターが環境ストレスに応答して、転写制御下の遺伝子に対する誘導性を示すことを見いだし、本発明を完成するに至った。
【0005】
すなわち、本発明は、以下を包含する。
(1) 以下の(a)、(b)又は(c)のDNAを含む、環境ストレス応答性プロモーター。
(a) 配列番号1の塩基配列からなるDNA
(b) 配列番号1の塩基配列において1若しくは複数の塩基が欠失、置換若しくは付加された塩基配列からなり、かつ環境ストレス応答性プロモーターとして機能するDNA
(c) 配列番号1の塩基配列と相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ環境ストレス応答性プロモーターとして機能するDNA
(2) 上記環境ストレスが生物ストレス及び/又は無生物ストレスであることを特徴とする(1)記載のプロモーター。
(3) (1)のプロモーターを含む発現ベクター。
(4) (3)記載の発現ベクターに、さらに任意の遺伝子が組み込まれた発現ベクター。
(5) (3)又は(4)記載の発現ベクターを含む形質転換体。
(6) (3)又は(4)記載の発現ベクターを含むトランスジェニック植物。
(7) 植物が、植物体、植物器官、植物組織又は植物培養細胞である(6)記載のトランスジェニック植物。
(8) (1)のプロモーターと、当該プロモーターによる発現制御可能なかたちで組み込まれたストレス耐性関連遺伝子とを含む発現ベクターでトランスジェニック植物を作製する工程と、
作製したトランスジェニック植物を培養又は栽培する工程とを含む、ストレス耐性植物の製造方法。
(9) (1)記載のプロモーターと、当該プロモーターによる発現制御可能なかたちで組み込まれたレポーター遺伝子とを有する発現ベクターにより形質転換された形質転換体に、検査対象の化合物を接触させる工程と、
上記形質転換体における上記レポーター遺伝子の発現を検出する工程と
を含む、生物ストレス及び/又は無生物ストレスに関与する化合物のスクリーニング方法。
(10) 上記化合物を接触させる工程では、生物ストレス及び/又は無生物ストレスを上記形質転換体に対して負荷することを特徴とする(9)記載のスクリーニング方法。
【発明の効果】
【0006】
本発明によれば、環境ストレス、特に生物ストレス及び/又は無生物ストレスに対する誘導性を示す、全く新規なプロモーターを提供することができる。また、本発明によれば、環境ストレス、特に生物ストレス及び/又は無生物ストレスに対する応答に関与する化合物を同定するための、全く新規なスクリーニング方法を提供することができる。
【発明を実施するための最良の形態】
【0007】
以下、本発明を詳細に説明する。
1.プロモーター
本発明に係るプロモーターは、以下の(a)、(b)又は(c)のDNAを含むものである
(a) 配列番号1の塩基配列からなるDNA
(b) 配列番号1の塩基配列において1若しくは複数の塩基が欠失、置換若しくは付加された塩基配列からなり、かつ環境ストレス応答性プロモーターとして機能するDNA
(c) 配列番号1の塩基配列と相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ環境ストレス応答性プロモーターとして機能するDNA
【0008】
ここで、配列番号1の塩基配列は、シロイヌナズナにおけるRAFL04-16-M08(At3g56710)におけるプロモーター領域を含む塩基配列である。RAFL04-16-M08は、sigA binding proteinをコードする遺伝子である。すなわち、配列番号1の塩基配列は、シロイヌナズナにおけるsigA binding protein遺伝子の開始コドン上流1500塩基からなる。
【0009】
また、上記(b)において、「複数の塩基」とは、1〜10個の塩基、好ましくは1〜5個の塩基を意味する。また、配列番号1の塩基配列において、1若しくは複数の塩基が欠失、置換若しくは付加され得る領域としては、特に限定されないが、配列番号1の塩基配列に含まれる、転写に関与するモチーフ配列を除く領域であることが好ましい。モチーフ配列としては、例えば、G-box、DRE、ABRE、GCC-box、W-box、TGA-box、P-box、MYB及びMYCを挙げることができる。配列番号1の塩基配列におけるこれらのモチーフ配列の存在位置を、図3に示す。図3では各モチーフ配列を下線で示している。
【0010】
さらに上記(c)において、ストリンジェントな条件とは、ナトリウム濃度が25〜500mM、好ましくは25〜300mMであり、温度が42〜68℃、好ましくは42〜65℃である。より具体的には、5×SSC(83mM NaCl、83mMクエン酸ナトリウム)、温度42℃である。
【0011】
また、本発明において、環境ストレスとは、具体的に生物ストレス及び/又は無生物ストレスを意味する。より具体的には、生物ストレスとは、カビ、細菌等が植物に感染することによって当該植物に対して負荷するストレスを意味する。無生物ストレスとは、低温度環境、高温度環境、乾燥環境、高塩濃度環境、高浸透圧環境等に植物を曝すことによって当該植物に対して負荷するストレスを意味する。
【0012】
上記(b)および(c)に示したDNAを含むプロモーターが環境ストレスに対して応答性を示すか否かは、当該プロモーターの下流にGUS遺伝子等のリポーター遺伝子を組み込んだ発現ベクターを用いて形質転換体を作製し、当該形質転換体が環境ストレスに曝された状態で上記リポーター遺伝子を発現しているか否かを検討することによって確認することができる。
【0013】
具体的に環境ストレスとしては、サリチル酸で処理する方法、プロベナゾールで処理する方法、バイオン、パラコート、重金属で処理する方法を挙げることができる。また、リポーター遺伝子としてGUS遺伝子を使用した場合には、環境ストレスに曝した後の形質転換植物におけるGUS活性を測定する。このような条件で環境ストレスに曝すことで、供試プロモーターが環境ストレスに対して応答性を示すか否かを判定することができる。
【0014】
2.発現ベクター
本発明の発現ベクターは、適当なベクターに本発明のプロモーターを連結(挿入)することにより得ることができる。本発明のプロモーターを挿入するためのベクターは、宿主中で複製可能なものであれば特に限定されず、例えばプラスミド、シャトルベクター、ヘルパープラスミドなどが挙げられる。
【0015】
プラスミドDNAとしては、大腸菌由来のプラスミド(例えばpBR322、pBR325、pUC118、pUC119、pUC18、pUC19、pBluescript等)、枯草菌由来のプラスミド(例えばpUB110、pTP5等)、酵母由来のプラスミド(例えばYEp13、YCp50等)などが挙げられ、ファージDNAとしてはλファージ(Charon4A、Charon21A、EMBL3、EMBL4、λgt10、λgt11、λZAP等)が挙げられる。さらに、レトロウイルス又はワクシニアウイルスなどの動物ウイルス、バキュロウイルスなどの昆虫ウイルスベクターを用いることもできる。
【0016】
ベクターに本発明のプロモーターを挿入するには、まず、精製されたDNAを適当な制限酵素で切断し、適当なベクター DNAの制限酵素部位又はマルチクローニングサイトに挿入してベクターに連結する方法などが採用される。
【0017】
本発明においては、任意遺伝子を発現させるため、上記発現ベクターに、さらに当該任意遺伝子を挿入することができる。任意の遺伝子を挿入する手法は、ベクターにプロモーターを挿入する方法と同様である。
【0018】
本発明のプロモーターは、その3'末端にレポーター遺伝子、例えば、植物で広く用いられているGUS遺伝子を連結して用いれば、GUS活性を調べることでプロモーターの強さを容易に評価することができる。なお、レポーター遺伝子としては、GUS遺伝子以外にも、ルシフェラーゼ、グリーンフルオレセイントプロテインなども用いることができる。
【0019】
このように、本発明においては、様々なベクターを用いることができる。さらに、本発明のプロモーターに目的の任意遺伝子をセンス又はアンチセンス方向で接続したものを作製し、これをバイナリーベクターと呼ばれるpBI101(Clonetech社)などのベクターに挿入することができる。
【0020】
3.形質転換体の作製
本発明の形質転換体は、本発明の発現ベクターを宿主中に導入することにより得ることができる。ここで、宿主としては、上述したプロモーターが機能しうるものであれば特に限定されるものではないが、植物が好ましい。宿主が植物である場合は、形質転換植物(トランスジェニック植物)は以下のようにして得ることができる。
【0021】
本発明において形質転換の対象となる植物は、植物体全体、植物器官(例えば葉、花弁、茎、根、種子等)、植物組織(例えば表皮、師部、柔組織、木部、維管束等)又は植物培養細胞のいずれをも意味するものである。形質転換に用いられる植物としては、アブラナ科、イネ科、ナス科、マメ科等に属する植物(下記参照)が挙げられるが、これらの植物に限定されるものではない。
アブラナ科:シロイヌナズナ(Arabidopsis thaliana)
ナス科:タバコ(Nicotiana tabacum)
イネ科:トウモロコシ(Zea mays) 、イネ(Oryza sativa)
マメ科:ダイズ(Glycine max)
【0022】
上記発現ベクターは、通常の形質転換方法、例えば電気穿孔法(エレクトロポレーション法)、アグロバクテリウム法、パーティクルガン法、PEG法等によって植物中に導入することができる。
【0023】
例えばエレクトロポレーション法を用いる場合は、パルスコントローラーを備えたエレクトロポレーション装置により、電圧500〜1600V、25〜1000μF、20〜30msecの条件で処理し、遺伝子を宿主に導入する。
【0024】
また、パーティクルガン法を用いる場合は、植物体、植物器官、植物組織自体をそのまま使用してもよく、切片を調製した後に使用してもよく、プロトプラストを調製して使用してもよい。このように調製した試料を遺伝子導入装置(例えばBio-Rad社のPDS-1000/He等)を用いて処理することができる。処理条件は植物又は試料により異なるが、通常は1000〜1800psi程度の圧力、5〜6cm程度の距離で行う。
【0025】
また、植物ウイルスをベクターとして利用することによって、本発明に係るプロモーターの発現制御下に目的遺伝子を植物体に導入することができる。利用可能な植物ウイルスとしては、例えば、カリフラワーモザイクウイルスが挙げられる。すなわち、まず、ウイルスゲノムを大腸菌由来のベクターなどに挿入して組換え体を調製した後、ウイルスのゲノム中に、これらの目的遺伝子を挿入する。このようにして修飾されたウイルスゲノムを制限酵素によって組換え体から切り出し、植物宿主に接種することによって、目的遺伝子を植物宿主に導入することができる。
【0026】
アグロバクテリウムのTiプラスミドを利用する方法においては、アグロバクテリウム(Agrobacterium)属に属する細菌が植物に感染すると、それが有するプラスミドDNAの一部を植物ゲノム中に移行させるという性質を利用して、本発明に係るプロモーター及び目的遺伝子を植物宿主に導入する。アグロバクテリウム属に属する細菌のうちアグロバクテリウム・ツメファシエンス(Agrobacterium tumefaciens)は、植物に感染してクラウンゴールと呼ばれる腫瘍を形成し、また、アグロバクテリウム・リゾゲネス(Agrobacteriumu rhizogenes)は、植物に感染して毛状根を発生させる。これらは、感染の際にTiプラスミド又はRiプラスミドと呼ばれる各々の細菌中に存在するプラスミド上のT-DNA領域(Transferred DNA)と呼ばれる領域が植物中に移行し、植物のゲノム中に組み込まれることに起因するものである。
【0027】
Ti又はRiプラスミド上のT-DNA領域中に、植物ゲノム中に組み込みたいDNAを挿入しておけば、アグロバクテリウム属の細菌が植物宿主に感染する際に目的とするDNAを植物ゲノム中に組込むことができる。
【0028】
形質転換の結果得られる腫瘍組織やシュート、毛状根などは、そのまま細胞培養、組織培養又は器官培養に用いることが可能であり、また従来知られている植物組織培養法を用い、適当な濃度の植物ホルモン(オーキシン、サイトカイニン、ジベレリン、アブシジン酸、エチレン、ブラシノライド等)の投与などにより植物体に再生させることができる。
【0029】
ここで、本発明に係るプロモーターによる発現制御下で導入される目的遺伝子としては、特に限定されないが、例えば、植物のストレス耐性に関連する遺伝子(ストレス耐性関連遺伝子と呼ぶ)を挙げることができる。ストレス耐性関連遺伝子としては、例えば、抗菌性タンパク質をコードするキチナーゼ遺伝子、β-1,3-グルカナーゼ遺伝子、さらには、ストレス耐性関連遺伝子の発現を制御する転写因子WRKY、ERF等を挙げることができる。これらのなかから選ばれる少なくとも1種のストレス耐性関連遺伝子を、本発明に係るプロモーターにより発現制御可能となるように宿主に導入することによって、ストレス耐性形質転換体を作製することができる。宿主として植物を使用した場合には、ストレス耐性植物を製造することができる。
【0030】
ところで、本発明に係るプロモーターと、当該プロモーターによる発現制御可能なかたちで組み込まれたレポーター遺伝子とを有する発現ベクターにより形質転換された形質転換体に検査対象の化合物を接触させ、形質転換体におけるレポーター遺伝子の発現を検出することによって、生物ストレス及び/又は無生物ストレスに関与する化合物をスクリーニングすることができる。ここで、検査対象の化合物を形質転換体に接触させる際には、生物ストレス及び/又は無生物ストレスを負荷した状態であってもよい。
【0031】
本発明の発現ベクターは、上記植物宿主に導入するのみならず、大腸菌(Escherichia coli)等のエッシェリヒア属、バチルス・ズブチリス(Bacillus subtilis)等のバチルス属、又はシュードモナス・プチダ(Pseudomonas putida)等のシュードモナス属に属する細菌、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)、シゾサッカロミセス・ポンベ(Schizosaccharomyces pombe)等の酵母、COS細胞、CHO細胞等の動物細胞、あるいはSf9等の昆虫細胞などに導入して形質転換体を得ることもできる。大腸菌、酵母等の細菌を宿主とする場合は、本発明の発現ベクターが該細菌中で自律複製可能であると同時に、本発明のプロモーター、リボソーム結合配列、目的遺伝子、転写終結配列により構成されていることが好ましい。また、プロモーターを制御する遺伝子が含まれていてもよい。
【0032】
細菌への組換えベクターの導入方法は、細菌にDNAを導入する方法であれば特に限定されるものではない。例えばカルシウムイオンを用いる方法、エレクトロポレーション法等が挙げられる。
【0033】
酵母を宿主とする場合は、例えばサッカロミセス・セレビシエ(Saccharomyces cerevisiae)、シゾサッカロミセス・ポンベ(Schizosaccharomyces pombe)などが用いられる。酵母への組換えベクターの導入方法は、酵母にDNAを導入する方法であれば特に限定されず、例えばエレクトロポレーション法、スフェロプラスト法、酢酸リチウム法等が挙げられる。
【0034】
動物細胞を宿主とする場合は、サル細胞COS-7、Vero、チャイニーズハムスター卵巣細胞(CHO細胞)、マウスL細胞などが用いられる。動物細胞への組換えベクターの導入方法としては、例えばエレクトロポレーション法、リン酸カルシウム法、リポフェクション法等が挙げられる。
【0035】
昆虫細胞を宿主とする場合は、Sf9細胞などが用いられる。昆虫細胞への組換えベクターの導入方法としては、例えばリン酸カルシウム法、リポフェクション法、エレクトロポレーション法などが挙げられる。
【0036】
遺伝子が宿主に組み込まれたか否かの確認は、PCR法、サザンハイブリダイゼーション法、ノーザンハイブリダイゼーション法等により行うことができる。例えば、形質転換体からDNAを調製し、DNA特異的プライマーを設計してPCRを行う。PCRは、前記プラスミドを調製するために使用した条件と同様の条件で行われる。その後は、増幅産物についてアガロースゲル電気泳動、ポリアクリルアミドゲル電気泳動又はキャピラリー電気泳動等を行い、臭化エチジウム、SYBR Green液等により染色し、そして増幅産物を1本のバンドとして検出することにより、形質転換されたことを確認する。また、予め蛍光色素等により標識したプライマーを用いてPCRを行い、増幅産物を検出することもできる。さらに、マイクロプレート等の固相に増幅産物を結合させ、蛍光又は酵素反応等により増幅産物を確認する方法も採用してもよい。
【0037】
4.植物の製造
本発明においては、上記形質転換植物細胞等から形質転換植物体に再生することができる。再生方法としては、カルス状の形質転換細胞をホルモンの種類、濃度を変えた培地へ移して培養し、不定胚を形成させ、完全な植物体を得る方法が採用される。使用する培地としては、LS培地、MS培地などが例示される。
【0038】
本発明の「植物体を製造する方法」は、上記植物プロモーターを挿入した植物発現ベクターを宿主細胞に導入して形質転換植物細胞を得て、該形質転換植物細胞から形質転換植物体を再生し、得られた形質転換植物体から植物種子を得て、該植物種子から植物体を生産する工程を含む。
【0039】
形質転換植物体から植物種子を得るには、例えば、形質転換植物体を発根培地から採取し、水を含んだ土を入れたポットに移植し、一定温度下で生育させて、花を形成させ、最終的に種子を形成させる。また、種子から植物体を生産するには、例えば、形質転換植物体上で形成された種子が成熟したところで、単離して、水を含んだ土に播種し、一定温度、照度下で生育させることにより、植物体を生産する。このようにして育種された植物は、導入されたプロモーターのストレス応答性に応じた環境ストレス耐性植物となる。
【実施例】
【0040】
以下、実施例を用いて本発明をより詳細に説明するが、本発明の技術的範囲は以下の実施例に限定されるものではない。
【0041】
〔実施例1〕マイクロアレイによる発現解析
本発明者らは、シロイヌナズナの約7,000個の完全長cDNAを有するマイクロアレイ(関ら、Plant J. 31, 279-292 (2002)に開示されたマイクロアレイ)を用いて、生物ストレス或いは無生物ストレスを負荷した条件下で特異的に発現している遺伝子を網羅的に解析した。本例で使用したマイクロアレイは、Arabidopsisの完全長cDNAライブラリーから単離した遺伝子に加えて、RD(Responsive to Dehydration)遺伝子、ERD(Early Responsive to Dehydration)遺伝子、内部標準としてλコントロール鋳型DNA断片(TX803、宝酒造株式会社製)、さらにネガティブコントロールとしてマウスのニコチン酸アセチルコリンレセプターのエプシロンサブユニット(nAChRE)遺伝子及びマウスのグルココルチコイドレセプターの相同性遺伝子からなる計約7000のcDNAを有している。
【0042】
このマイクロアレイを用いて、以下のように環境ストレスに対して応答性を示す遺伝子を解析した。実験に用いたシロイヌナズナは、2%Murashige and Skoog塩(和光純薬社製)、2%スクロース及び0.8%バクトアガー(Difco社製)を含むGM培地で発芽させ、その後、16時間(午前5時〜午後9時)を明下とし8時間を暗下とするサイクルで、22℃のチャンバー内で3週間成長させた。また、様々な環境ストレス処理は、ほぼ同時刻(午前9時〜午前10時の間)に開始した。
【0043】
本実験において、〔ストレス負荷時の発現量〕/〔ストレスがない時の発現量〕の値がλコントロール鋳型DNA断片と比較して3倍以上であり、且つ、少なくとも一回の実験で1000以上のシグナル値を示すものを環境ストレス負荷によって特異的に発現する遺伝子と認定した。本例においては、生物ストレス負荷時及び無生物ストレス負荷時において特異的に発現する遺伝子を解析した。
【0044】
生物ストレスとしては、非病原性糸状菌(Alternaria alternata O-276;鳥取大学の尾谷氏より供与)、不親和性糸状菌(Alternaria brassicicola O-264;鳥取大学の尾谷氏より供与)及び親和性糸状菌(Colletotrichum higginsianum MAFF 305635;MAFF Genebankより入手)を接種することによって行った。無生物ストレスは、乾燥処理、低温処理、高塩濃度処理、UV-C処理及びCuSO4処理によって行った。これら無生物ストレス及び生物ストレスは、“The cDNA Microarray Analysis Using an Arabidopsis pad3 Mutant Reveals the Expression Profiles and Classification of Genes Induced by Alternaria brassicicola Attack”(鳴坂らPlant Cell Physiol. 44(4) 377-387 (2003))に記載の方法に準じて行った。
【0045】
シロイヌナズナからのtotal RNAの抽出は、生物ストレス負荷から10、24及び48時間後に行い、無生物ストレス負荷から2、5、10及び24時間後に行った。また、コントロールとして、蒸留水処理から2、5、10、24及び48時間後の植物からもtotal RNAを抽出した。なお、total RNAの抽出にはTRIZOL Reagent(Invitrogen社製)を使用し、total RNAからのmRNAの抽出にはmRNA isolation kit(Miltenyi Biotec社製)を使用した。
【0046】
その結果、生物ストレス負荷によって900個の遺伝子が特異的に発現していることが判った。なお、Alternaria alternataの接種によって118個の遺伝子が特異的に発現し、Alternaria brassicicolaの接種によって864個の遺伝子が特異的に発現し、Colletotrichum higginsianumの接種によって142個の遺伝子が特異的に発現した。これに対して、無生物ストレス負荷によって1340個の遺伝子が特異的に発現していることが判った。なお、乾燥処理によって740個の遺伝子が特異的に発現し、低温処理によって438個の遺伝子が特異的に発現し、高塩濃度処理によって605個の遺伝子が特異的に発現し、UV-C処理によって290個の遺伝子が特異的に発現し、CuSO4処理によって676個の遺伝子が特異的に発現した。これらの結果を図1にまとめる。図1から判るように、生物ストレス及び無生物ストレスによって特異的に発現する遺伝子の数は469個であった。
【0047】
この469個の遺伝子のうち、RAFL04-16-M08については、サリチル酸処理特異的に発現していることが明らかとなった。生物ストレス及び無生物ストレスによるRAFL04-16-M08の発現量の変化を図2に示す。なお、図2において縦軸は規格化した強度、すなわち、発現量の増加を示し、横軸はストレス負荷からの時間を示している。
【0048】
なお、RAFL04-16-M08は、SigA binding protein(At3g56710)である。以下、RAFL04-16-M08を「SigA BP遺伝子」と称する。なお、実施例中、低温処理は、4℃で生育させる条件で行った。高塩濃度処理はシロイヌナズナを250mMのNaCl溶液で水耕栽培する条件で行った。UV-C処理はシロイヌナズナに1kJの紫外線を照射する条件で行った。CuSO4処理は10mMのCuSO4溶液を噴霧する条件で行った。アブシジン酸処理は100μMのABA溶液で水耕栽培する条件とした。サリチル酸処理は5mMのサリチル酸溶液を噴霧する条件で行った。ジャスモン酸処理は200μMのジャスモン酸メチル溶液で水耕栽培する条件とした。エチレン処理(エテフォン処理)は1mMのエテフォン溶液で水耕栽培する条件とした。パラコート処理は25μMのパラコート溶液を噴霧する条件で行った。ローズベンガル処理は20mMのローズベンガル溶液を噴霧する条件で行った。
【0049】
以上、本例によってSigA BP遺伝子は生物ストレス及び無生物ストレスのいずれを負荷しても発現が誘導されることが明らかとなり、当該SigA BP遺伝子の上流に存在するプロモーターが生物ストレス及び無生物ストレス誘導性のプロモーターであることが推測された。
【0050】
〔実施例2〕マイクロアレイによる発現解析2
本発明者らは、シロイヌナズナの約7,000個の完全長cDNAを有するマイクロアレイ(関ら、Plant J. 31, 279-292 (2002)に開示されたマイクロアレイ)を用いて、生物ストレス或いは無生物ストレスを負荷した条件下で特異的に発現している遺伝子を網羅的に解析した。本例で使用したマイクロアレイは、Arabidopsisの完全長cDNAライブラリーから単離した遺伝子に加えて、RD(Responsive to Dehydration)遺伝子、ERD(Early Responsive to Dehydration)遺伝子、内部標準としてλコントロール鋳型DNA断片(TX803、宝酒造株式会社製)、さらにネガティブコントロールとしてマウスのニコチン酸アセチルコリンレセプターのエプシロンサブユニット(nAChRE)遺伝子及びマウスのグルココルチコイドレセプターの相同性遺伝子からなる計約7000のcDNAを有している。
【0051】
このマイクロアレイを用いて、以下のように環境ストレスに対して応答性を示す遺伝子を解析した。植物材料として、土(ダイオ化成社製)に播種し、24時間の明暗サイクルを明時間16時間及び暗時間8時間として4週間栽培したシロイヌナズナ(生態型:コロンビア)を用いた。また、様々な環境ストレス処理は、ほぼ同時刻(午前9時〜午前10時の間)に開始した。
【0052】
本実験において、〔ストレス負荷時の発現量〕/〔ストレスがない時の発現量〕の値がλコントロール鋳型DNA断片と比較して3倍以上であり、且つ、少なくとも一回の実験で1000以上のシグナル値を示すものを環境ストレス負荷によって特異的に発現する遺伝子と認定した。本例においては、生物ストレス負荷時及び無生物ストレス負荷時において特異的に発現する遺伝子を解析した。
【0053】
生物ストレスとしては、非病原性糸状菌(Alternaria alternata O-276;鳥取大学の尾谷氏より供与)、不親和性糸状菌(Alternaria brassicicola O-264;鳥取大学の尾谷氏より供与)及び親和性糸状菌(Colletotrichum higginsianum MAFF 305635;MAFF Genebankより入手)を接種することによって行った。無生物ストレスは、乾燥処理、低温処理、高塩濃度処理、UV-C処理及びCuSO4処理によって行った。これら無生物ストレス及び生物ストレスは、“The cDNA Microarray Analysis Using an Arabidopsis pad3 Mutant Reveals the Expression Profiles and Classification of Genes Induced by Alternaria brassicicola Attack”(鳴坂らPlant Cell Physiol. 44(4) 377-387 (2003))に記載の方法に準じて行った。
【0054】
シロイヌナズナからのtotal RNAの抽出は、生物ストレス負荷から10、24及び48時間後に行い、無生物ストレス負荷から2、5、10及び24時間後に行った。また、コントロールとして、蒸留水処理から2、5、10、24及び48時間後の植物からもtotal RNAを抽出した。なお、total RNAの抽出にはTRIZOL Reagent(Invitrogen社製)を使用し、total RNAからのmRNAの抽出にはmRNA isolation kit(Miltenyi Biotec社製)を使用した。
【0055】
発現遺伝子のうち、RAFL04-16-M08については、サリチル酸処理特異的に発現していることが明らかとなった。生物ストレス及び無生物ストレスによるRAFL04-16-M08の発現量の変化を図2に示す。なお、図2において縦軸は規格化した強度、すなわち、発現量の増加を示し、横軸はストレス負荷からの時間を示している。
【0056】
サリチル酸処理は5mMのサリチル酸溶液を噴霧する条件で行った。ジャスモン酸処理は100μMのジャスモン酸メチル溶液を噴霧する条件で行った。エチレン処理(エテフォン処理)は1mMのエテフォン溶液を噴霧する条件で行った。パラコート処理は25μMのパラコート溶液を噴霧する条件で行った。
【0057】
以上、本例によってSigA BP遺伝子は生物ストレス及び無生物ストレスのいずれを負荷しても発現が誘導されることが明らかとなり、当該SigA BP遺伝子の上流に存在するプロモーターが生物ストレス及び無生物ストレス誘導性のプロモーターであることが推測された。
【0058】
〔実施例3〕SigA BP遺伝子のプロモーター領域の解析
本例では、生物ストレス及び無生物ストレス誘導性のプロモーターであると推測された、SigA BP遺伝子の上流に存在するプロモーターを単離し、そのプロモーター活性を解析した。
【0059】
先ず、SigA BP遺伝子のcDNAの塩基配列を定法に従って決定した。次に、この塩基配列情報に基づいてシロイヌナズナゲノム・データベース(URL; http://rarge.gsc.riken.go.jp/)を検索し、SigA BP遺伝子の開始コドンより上流約1.5kbをプロモーターとして同定した。シロイヌナズナにおいては、開始コドンより上流約1.0kb内にプロモーターが存在し、開始コドンより約150b内に5’非翻訳領域が存在することが知られているためである(MaleckらNature Genet. 26, 403-410 (2000))。
【0060】
本例で同定したプロモーター領域の塩基配列を配列番号1及び図3に示す。なお、図3には、塩基配列情報に基づいて特定されたシスエレメント領域を、下線で示している。また、下線の下に付記した文字はシスエレメントの種類を示している。図3中「W-box」はWRKY転写因子の結合領域を示している。WRKY転写因子は、病害防御、老化及びトライコーム形成等の様々な生理学的問題の制御に関わっており(EulgemらTrends Plant Sci. 5, 199-206 (2000))、生物ストレスに対する応答性に寄与するものと考えられた。図3中「ACGT」及び「TGA-box」はactivation sequence-1 (as-1)因子のコア配列(TGACGT/C)を示している(Lam et al.,(1989)Proc. Natl. Acad. Sci. USA 86, 7890-7894)。なおas-1因子は酸化ストレス応答性因子として作用する。「ACGT」及び「TGA-box」は、活性酸素種応答性プロモーター内においてシスエレメントとして作用すると考えられる(GarretonらPlant Physiol. 130, 1516-1526 (2002))。図3中の「G-box」はサリチル酸(Maleck, K. et al (2000) Nature Genet. 26, 403-410.)、アブシジン酸’Guiltinan, M.J. et al. (1990) Science 250, 267-271.)、エチレン(Eyal, Y. et al. (1993) Plant J. 4, 225-234.)及びジャスモン酸(Mason, H.S. et al. (1993) Plant Cell 5, 241-251.)に応答するシスエレメント(ACGTG)として知られている。図3中の「P-box」は病害防御応答に関するシスエレメント(CCA/TA/TCC)して作用する。
【0061】
次に、配列番号1に示した塩基配列からなるプロモーターが無生物ストレス及び/又は生物ストレスに対して誘導性を示すか否かを検討した。配列番号1に示した塩基配列からなるプロモーター及び当該プロモーターの下流に位置するGUS遺伝子を有するベクターを以下のように作製した。
【0062】
シロイヌナズナゲノムDNAからPCRを用いて、SigA BP遺伝子の開始コドンより上流1.5kbをSigA BP遺伝子のプロモーターとしてクローニングした。PCRはプライマー配列に制限酵素Hind IIIの認識部位を付与したプライマー(プライマー1:cccaagcttccattcgtttcttaatccttacc(配列番号2))とBamHIの認識部位を付与したプライマー(プライマー2:cgggatccgttcggttcctgtaagagaattcag(配列番号3))を使用して、最初に94℃で2分反応させた後、94℃で30秒、55℃で30秒及び74℃で2分のサイクルを25サイクル、最後に74℃で7分の反応条件で行った。
【0063】
このSigA BP遺伝子のプロモーターDNAを制限酵素Hind IIIとBamHIで切断し、バイナリーベクターと呼ばれるpBI101(Clonetech社)ベクター DNAのマルチクローニングサイト、制限酵素Hind IIIとBamHI切断部位に挿入してベクターに連結した。これによりSigA BP遺伝子のプロモーターの3'末端にレポーター遺伝子、GUS遺伝子を連結した、ベクターを構築した。
【0064】
〔実施例4〕プロモーター活性の検定
このように作製したベクターを用いてアグロバクテリウムを介してシロイヌナズナを形質転換し、最終的に形質転換シロイヌナズナホモ接合体株を得た。得られたホモ接合体株を播種後25〜28日経過した時点で、マイクロアレイ解析によってSigA BP遺伝子を特異的に誘導することがあきらかとなったサリチル酸で処理した後、GUS活性を測定することによって、プロモーター活性を評価した。具体的には、0.5mMのサリチル酸またはコントロールとして蒸留水10mlをシロイヌナズナ形質転換体に散布した。散布24時間後にロゼット葉1〜2枚程度を1.5ml チューブに入れ、200μlの抽出用緩衝液(50mM リン酸ナトリウム緩衝液pH7.0, 10mM EDTA, 0.1%Triton X-100, 10mM 2-メルカプトエタノール)を添加し、氷上でホモジナイザーにて破砕した。
【0065】
15000rpm, 4℃,で10分間遠心した後、上清10μlを2mlチューブに移し、基質として100μlの4-methylumbelliferyl β-D-glucuronide (4-MUG)(0.4mg/ml抽出用緩衝液)を加え、37℃で1時間反応した。1時間後、1.4mlの0.2M Na2CO3を添加して反応を停止した。その後、365nmの励起光による455nmの放出スペクトルを測定することにより、単位時間、単位タンパク質含量当たりの4-MUの生成量(pmol 4-MU/min/mg protein)を求めた。上清中のタンパク質含量はBioRad protein assay Kitを使用して、BSAをスタンダードとして、添付のマニュアルにしたがって定量した。
【0066】
その結果、図4に示すように、蒸留水噴霧処理と比較して、サリチル酸処理によってGUS活性は増強していることが判った。なお図4において、活性値は3サンプルの平均±標準偏差で示しており、各棒グラフ上の数値は水処理でのGUS活性を1.0としたときの各薬剤処理でのGUS活性の相対比を表している。
【0067】
〔実施例5〕PR-1プロモーターとの比較検定-1
0.5mMのサリチル酸、0.5mMのプロベナゾールまたはコントロールとして蒸留水10mlを 実施例4と同様に栽培したSigA BP::GUSおよびPR-1::GUS形質転換シロイヌナズナ(いずれもホモ接合体株)の株元に滴下した。処理2時間後に実施例4で示した方法に従ってGUS活性を測定した。その結果、図5に示すように、SigA BP::GUS形質転換シロイヌナズナでは蒸留水噴霧処理と比較して、サリチル酸およびプロベナゾール処理によってGUS活性は増強していることが判った。一方、PR-1::GUS形質転換シロイヌナズナではGUS活性の増強は認められなかった。新規なplant activatorを見出すには多数の試験化合物を効率よく検定することが重要である。本実施例で示されたように、SigA BPの発現を指標とすることによって、従来のマーカー遺伝子(PR-1)に比較して短時間で化合物の検定を行うことが可能となる。なお図5において、活性値は3サンプルの平均±標準偏差で示しており、各棒グラフ上の数値は水処理でのGUS活性を1.0としたときの各薬剤処理でのGUS活性の相対比を表している。
【0068】
本実施例の結果は図6に示したサリチル酸処理後のSigA BP およびPR-1遺伝子のマイクロアレイ解析による発現の経時変化からも支持される。すなわち、SigA BPはサリチル酸処理2時間後には遺伝子発現が認められているのに対して、PR-1は有意な遺伝子発現が認められていない。
【0069】
なお、PR-1::GUS形質転換シロイヌナズナは以下の方法で構築したベクターを用いてアグロバクテリウムを介して作出した。
【0070】
シロイヌナズナゲノムDNAからPCRを用いて、PR-1遺伝子の開始コドンより上流1290bpをPR-1遺伝子のプロモーターとしてクローニングした。PCRはプライマー配列に制限酵素Hind IIIの認識部位を付与したプライマー(プライマー1:gcacaagcttgttttaac(配列番号4))とXbaIの認識部位を付与したプライマー(プライマー2:gagatctagattttctaagttgataatggt(配列番号5))を使用して、最初に94℃で2分反応させた後、94℃で30秒、55℃で30秒及び74℃で1分のサイクルを30サイクル、最後に74℃で7分の反応条件で行った。
【0071】
このPR-1遺伝子のプロモーターDNAを制限酵素Hind IIIとXbaIで切断し、バイナリーベクターと呼ばれるpBI101(Clonetech社)ベクター DNAのマルチクローニングサイト、制限酵素Hind IIIとXbaI切断部位に挿入してベクターに連結した。これによりPR-1遺伝子のプロモーターの3'末端にレポーター遺伝子、GUS遺伝子を連結した、ベクターを構築した。
【0072】
〔実施例6〕PR-1プロモーターとの比較検定-2
0.1mMおよび0.05 mM のプロベナゾールまたはコントロールとして蒸留水10mlを実施例4と同様に栽培したSigA BP::GUS形質転換シロイヌナズナ(ホモ接合体株)の株元に滴下した。処理1日後に実施例4で示した方法に従ってGUS活性を測定した。その結果、図7に示すように、SigA BP::GUS形質転換シロイヌナズナでは蒸留水噴霧処理と比較して、0.05mM プロベナゾール処理でもGUS活性は増強していることが判った。一方、PR-1::GUS形質転換シロイヌナズナでは0.1 mM 処理ではGUS活性の増強が認められたが、0.05mM処理では認められなかった。本実施例で示されたように、SigA BPの発現を指標とすることによって従来のマーカー遺伝子(PR-1)では評価できないポテンシャルの低いplant activatorのリード化合物を見出すことが可能となる。なお、図7において活性値は3サンプルの平均±標準偏差で示しており、各棒グラフ上の数値は水処理でのGUS活性を1.0としたときの各薬剤処理でのGUS活性の相対比を表している。
【図面の簡単な説明】
【0073】
【図1】生物ストレス誘導性の遺伝子及び無生物ストレス誘導性の遺伝子をマイクロアレイで解析した結果を分類したベン図である。
【図2】生物ストレス誘導性及び無生物ストレスによるSigA BP遺伝子の発現量の変化を示す特性図である。
【図3】本発明に係るプロモーターの塩基配列を解析した結果を示す図である。
【図4】本発明に係るプロモーターの活性を、GUS遺伝子の発現に起因するGUS活性で測定した結果を示す特性図である。
【図5】本発明に係るプロモーターの活性を、PR-1プロモーターと比較した結果を示す特性図である。
【図6】サリチル酸散布によるSigA BPおよびPR-1遺伝子の発現量の変化を示す特性図である。
【図7】本発明に係るプロモーターの薬剤濃度に対する感受性を、PR-1プロモーターと比較した結果を示す特性図である。
【特許請求の範囲】
【請求項1】
以下の(a)、(b)又は(c)のDNAを含む、環境ストレス応答性プロモーター。
(a) 配列番号1の塩基配列からなるDNA
(b) 配列番号1の塩基配列において1若しくは複数の塩基が欠失、置換若しくは付加された塩基配列からなり、かつ環境ストレス応答性プロモーターとして機能するDNA
(c) 配列番号1の塩基配列と相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ環境ストレス応答性プロモーターとして機能するDNA
【請求項2】
上記環境ストレスが生物ストレス及び/又は無生物ストレスであることを特徴とする請求項1記載のプロモーター。
【請求項3】
請求項1のプロモーターを含む発現ベクター。
【請求項4】
請求項3記載の発現ベクターに、上記プロモーターによる発現制御可能なかたちで遺伝子が組み込まれた発現ベクター。
【請求項5】
請求項3又は請求項4記載の発現ベクターを含む形質転換体。
【請求項6】
請求項3又は請求項4記載の発現ベクターを含むトランスジェニック植物。
【請求項7】
植物が、植物体、植物器官、植物組織又は植物培養細胞である請求項6記載のトランスジェニック植物。
【請求項8】
請求項1のプロモーターと、当該プロモーターによる発現制御可能なかたちで組み込まれたストレス耐性関連遺伝子とを含む発現ベクターでトランスジェニック植物を作製する工程と、
作製したトランスジェニック植物を培養又は栽培する工程とを含む、ストレス耐性植物の製造方法。
【請求項9】
請求項1記載のプロモーターと、当該プロモーターによる発現制御可能なかたちで組み込まれたレポーター遺伝子とを有する発現ベクターにより形質転換された形質転換体に、検査対象の化合物を接触させる工程と、
上記形質転換体における上記レポーター遺伝子の発現を検出する工程と
を含む、生物ストレス及び/又は無生物ストレスに関与する化合物のスクリーニング方法。
【請求項10】
上記化合物を接触させる工程では、生物ストレス及び/又は無生物ストレスを上記形質転換体に対して負荷することを特徴とする請求項9記載のスクリーニング方法。
【請求項1】
以下の(a)、(b)又は(c)のDNAを含む、環境ストレス応答性プロモーター。
(a) 配列番号1の塩基配列からなるDNA
(b) 配列番号1の塩基配列において1若しくは複数の塩基が欠失、置換若しくは付加された塩基配列からなり、かつ環境ストレス応答性プロモーターとして機能するDNA
(c) 配列番号1の塩基配列と相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ環境ストレス応答性プロモーターとして機能するDNA
【請求項2】
上記環境ストレスが生物ストレス及び/又は無生物ストレスであることを特徴とする請求項1記載のプロモーター。
【請求項3】
請求項1のプロモーターを含む発現ベクター。
【請求項4】
請求項3記載の発現ベクターに、上記プロモーターによる発現制御可能なかたちで遺伝子が組み込まれた発現ベクター。
【請求項5】
請求項3又は請求項4記載の発現ベクターを含む形質転換体。
【請求項6】
請求項3又は請求項4記載の発現ベクターを含むトランスジェニック植物。
【請求項7】
植物が、植物体、植物器官、植物組織又は植物培養細胞である請求項6記載のトランスジェニック植物。
【請求項8】
請求項1のプロモーターと、当該プロモーターによる発現制御可能なかたちで組み込まれたストレス耐性関連遺伝子とを含む発現ベクターでトランスジェニック植物を作製する工程と、
作製したトランスジェニック植物を培養又は栽培する工程とを含む、ストレス耐性植物の製造方法。
【請求項9】
請求項1記載のプロモーターと、当該プロモーターによる発現制御可能なかたちで組み込まれたレポーター遺伝子とを有する発現ベクターにより形質転換された形質転換体に、検査対象の化合物を接触させる工程と、
上記形質転換体における上記レポーター遺伝子の発現を検出する工程と
を含む、生物ストレス及び/又は無生物ストレスに関与する化合物のスクリーニング方法。
【請求項10】
上記化合物を接触させる工程では、生物ストレス及び/又は無生物ストレスを上記形質転換体に対して負荷することを特徴とする請求項9記載のスクリーニング方法。
【図1】


【図2】
【図3】

【図4】
【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2007−202461(P2007−202461A)
【公開日】平成19年8月16日(2007.8.16)
【国際特許分類】
【出願番号】特願2006−24536(P2006−24536)
【出願日】平成18年2月1日(2006.2.1)
【出願人】(000000169)クミアイ化学工業株式会社 (86)
【Fターム(参考)】
【公開日】平成19年8月16日(2007.8.16)
【国際特許分類】
【出願日】平成18年2月1日(2006.2.1)
【出願人】(000000169)クミアイ化学工業株式会社 (86)
【Fターム(参考)】
[ Back to top ]