
新規レポーター遺伝子を用いたスクリーニング方法
【課題】種々の抗菌作用を付加した際に特異的に発現する遺伝子を特定し、当該遺伝子をレポーター遺伝子として用いることで、公知の抗菌剤の候補物質或いは公知の抗菌剤と同等の抗菌作用を有する物質をスクリーニングする。
【解決手段】被検物質を作用させた細胞における、特定の遺伝子の発現を測定する工程と、上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を抗菌剤の候補物質として同定する工程とを含む。
【解決手段】被検物質を作用させた細胞における、特定の遺伝子の発現を測定する工程と、上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を抗菌剤の候補物質として同定する工程とを含む。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、作用物質による刺激により特異的に発現する遺伝子をレポーター遺伝子として使用するスクリーニング方法及び当該遺伝子の発現を制御するプロモータに関する。
【背景技術】
【0002】
微生物は、高浸透圧、高温、栄養飢餓、酸化、創傷といった様々な環境ストレスに曝される。これら環境ストレスに微生物が曝されると、特定の遺伝子発現といった生化学的、生理学的及び分子的な反応が誘導され、環境ストレスに対する応答性及び順応性を示すことが知られている。このような反応は、微生物細胞内における刺激の認識及びシグナル伝達の結果として進行する。抗菌剤への暴露も環境ストレスの1つであり、特定の遺伝子発現が誘導されることが示唆されるが、抗菌剤の抗菌作用と、当該抗菌作用の付加時に特異的に発現誘導される遺伝子との関係は多くの知見がないのが現状である。
【0003】
例えば、呼吸は生物にとって生存に必須であり、呼吸鎖阻害剤は抗菌剤として高い効果を示す。したがって、公知の呼吸鎖阻害剤と同様の抗菌作用を示す物質について、更なる開発が望まれている。
【0004】
また、エルゴステロールは真菌に特有の細胞膜の成分で、生存に必須であり、エルゴステロール生合成阻害剤は抗菌剤として高い効果を示す。したがって、公知のエルゴステロール生合成阻害剤と同様の抗菌作用を示す物質について、更なる開発が望まれている。
【0005】
さらに、グルカン、キチンなどから成る細胞壁は真菌に特有の成分で、生存に必須であり、細胞壁生合成阻害剤は抗菌剤として高い効果を示す。したがって、公知の細胞壁生合成阻害剤と同様の抗菌作用を示す物質について、更なる開発が望まれている。
【0006】
さらにまた、cell wall integrity経路は、BckA(MAPKKK)→MkkA(MAPKK)→MpkA(MAPK)からなるMAP キナーゼシグナル伝達経路を中心とした細胞壁構築制御経路であり、真菌類に広く保存されている生体システムであり、細胞壁生合成阻害剤への暴露などにより細胞壁に損傷を受けると本経路が活性化し、細胞壁生合成関連遺伝子の転写を制御して細胞壁の再構築が行われることが知られている。cell wall integrity経路の遮断または構成的な活性化は細胞に重篤な障害を引き起こすことから、cell wall integrity経路阻害薬は抗菌剤として高い効果を示すと考えられる。したがって、cell wall integrity経路阻害薬の開発が望まれている。
【0007】
さらにまた、MpkB経路は、出芽酵母のSte11(MAPKKK)→Ste7(MAPKK)→Fus3/Kss1(MAPK)からなるMAP キナーゼシグナル伝達経路の相同経路で、真菌類に広く保存されている生体システムであり、多くの植物病原菌において、本経路が病原性に必須であることが報告されていることから、MpkB経路阻害薬は抗菌剤として高い効果を示すと考えられる。したがって、MpkB経路阻害薬の開発が望まれている。
【0008】
さらにまた、HOG経路は、出芽酵母のSsk2/22(MAPKKK)→Pbs2(MAPKK)→Hog1(MAPK)からなるMAP キナーゼシグナル伝達経路の相同経路で、真菌類に広く保存されている生体システムであり、多くの植物病原菌において、本経路が病原性に必須であることが報告されている。HOG経路の上流には、ヒスチジンキナーゼ→リン酸基仲介因子→レスポンスレギュレーターからなる二成分性情報伝達系が存在し、このヒスチジンキナーゼに作用してHOG経路を構成的に活性化することで抗菌活性を示す薬剤が農薬の有効成分として実用化もされており、HOG経路作動薬は抗菌剤として高い効果を示す。したがって、公知のHOG経路作動薬と同様の抗菌作用を示す物質について、更なる開発が望まれていることから、本経路を標的とする薬剤の開発が望まれている。
【発明の概要】
【発明が解決しようとする課題】
【0009】
そこで、本発明は、上述したような実情に鑑み、種々の抗菌作用を付加した際に特異的に発現する遺伝子を特定し、当該遺伝子をレポーター遺伝子として用いることで、公知の抗菌剤の候補物質或いは公知の抗菌剤と同等の抗菌作用を有する物質をスクリーニングする方法を提供することを目的とする。
【0010】
また、本発明は、種々の抗菌作用を付加した際に特異的に発現する遺伝子及び当該遺伝子の発現を制御するプロモータ領域を特定し、種々の抗菌作用を付加したときに発現を誘導するといった特徴を有するプロモータを提供することを目的とする。
【課題を解決するための手段】
【0011】
上述した目的を達成した本発明は以下を包含する。
本発明に係るスクリーニング方法は、被検物質を作用させた細胞における、表1に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる少なくとも1種の遺伝子の発現を測定する工程と、上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を呼吸鎖阻害剤の候補物質として同定する工程とを含んでいる。
【0012】
【表1】

【0013】
また、本発明に係るスクリーニング方法は、被検物質を作用させた細胞における、表2に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる少なくとも1種の遺伝子の発現を測定する工程と、上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質をエルゴステロール生合成阻害剤の候補物質として同定する工程とを含んでいる。
【0014】
【表2】
【0015】
さらに、本発明に係るスクリーニング方法は、被検物質を作用させた細胞における、表3に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる少なくとも1種の遺伝子の発現を測定する工程と、上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を細胞壁生合成阻害剤の候補物質として同定する工程とを含んでいる。
【0016】
【表3】
【0017】
さらにまた、本発明に係るスクリーニング方法は、被検物質を作用させた細胞における、スーパーオキシドデスムターゼ遺伝子(Genbankアクセッション番号:XM_653297、配列番号71)の発現を測定する工程と、上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質をMpkB経路阻害剤の候補物質として同定する工程とを含んでいる。
【0018】
さらにまた、本発明に係るスクリーニング方法は、被検物質を作用させた細胞における、表4に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる少なくとも1種の遺伝子の発現を測定する工程と、上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を細胞壁構築(cell wall integrity)経路阻害剤の候補物質として同定する工程とを含んでいる。
【0019】
【表4】
【0020】
さらにまた、被検物質を作用させた細胞における、agsA遺伝子(Genbankアクセッション番号:XM_658397、配列番号83)及び/又はagsB遺伝子(Genbankアクセッション番号:XM_655819、配列番号84)の発現を測定する工程と、上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を細胞壁構築(cell wall integrity)経路阻害剤の候補物質として同定する工程とを含んでいる。
【0021】
さらにまた、本発明に係るスクリーニング方法は、被検物質を作用させた細胞における、cat1遺伝子(Genbankアクセッション番号:XM_365841、配列番号85)の発現を測定する工程と、上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を浸透圧応答(HOG: high osmolarity glycerol)経路活性化剤の候補物質として同定する工程とを含んでいる。
【発明の効果】
【0022】
以上のように、本発明に係るスクリーニング方法によれば、呼吸鎖阻害剤の候補物質、エルゴステロール生合成阻害剤の候補物質、細胞壁生合成阻害剤の候補物質、MpkB経路阻害剤の候補物質、細胞壁構築(cell wall integrity)経路阻害剤の候補物質又は浸透圧応答(HOG: high osmolarity glycerol)経路活性化剤の候補物質を同定することができる。すなわち、本発明によれば、上記の各種抗菌作用に対する簡便な評価系を確立することができ、多数の化合物群からなる薬剤ライブラリーから候補物質を、高速かつ高効率で同定することができる。
【図面の簡単な説明】
【0023】
【図1】シアン耐性呼吸酵素遺伝子(AOX)(上段)及びNADH-デヒドロゲナーゼ遺伝子(下段)についてRT-PCRにより測定した発現量をactin当たりの発現量として示す特性図である。
【図2】糸状菌形質転換ベクターpCH11の概略構成図である。
【図3】pCH11-EGFP-LacIの概略構成図である。
【図4】pCH11-PrAOX-EGFP-LacIプラスミドを導入したマグナポルテ・グリセアに対して、呼吸鎖阻害剤及び比較薬剤を処理した時のEGFPの蛍光を観察した結果を示す特性図である。
【図5】Cytochrome P450ステロール14αジメチラーゼ(MGG_04628、上段)及びステロール24Cメチルトランスフェラーゼ(MGG_10860、下段)についてRT-PCRにより測定した発現量をactin当たりの発現量として示す特性図である。
【図6】pCH11- Cyp51-EGFP-LacIプラスミドを導入したマグナポルテ・グリセアに対して、エルゴステロール生合成阻害剤及び比較薬剤を処理した時のEGFPの蛍光を観察した結果を示す特性図である。
【図7】MGG_02004(上段の左)、MGG_00692(上段の右)、MGG_04943(中段の左)、MGG_09639(中段の右)、MGG_05133(下段の左)及びMGG_02773(下段の右)についてRT-PCRにより測定した発現量をactin当たりの発現量として示す特性図である。
【図8】pNA(N)EGFPの概略構成図である。
【図9】pMSTU1-EGFP及びpAGS1-EGFPを導入したマグナポルテ・グリセアに対して、細胞壁生合成阻害剤を処理した時のEGFPの蛍光を観察した結果を示す特性図である。
【図10】mpkBΔ株におけるsodM遺伝子の発現量をHistone H2B当たりの発現量として示す特性図である。
【図11】PagsB-EGFP形質転換体及びPagsA-EGFP形質転換体について、蛍光を観察した結果を示す特性図である。
【図12】いもち病菌におけるcat1遺伝子についてRT-PCRにより測定した発現量をβ-tubulin当たりの発現量として示す特性図である。
【発明を実施するための形態】
【0024】
以下、本発明をより詳細に説明する。
本発明に係るスクリーニング方法は、種々の被検物質の中から、呼吸鎖阻害剤の候補物質、エルゴステロール生合成阻害剤の候補物質、細胞壁生合成阻害剤の候補物質、MpkB経路阻害剤の候補物質、細胞壁構築(cell wall integrity)経路阻害剤の候補物質又は浸透圧応答(HOG: high osmolarity glycerol)経路活性化剤の候補物質を同定する方法である。ここで、被験物質としては、特に限定されず如何なる物質であってもよい。被験物質としては、単独の物質であってもよいし、複数の構成成分からなる混合物であってもよい。被験物質としては、例えば植物からの抽出物のように未同定の物質を含むような構成であってもよいし、既知の組成物を所定の組成比で含むような構成であってもよい。また、被験物質としては、タンパク質、核酸、脂質、多糖類、有機化合物及び無機化合物のいずれでもよい。
【0025】
また、本明細書において、呼吸鎖阻害剤とは、微生物、より好ましくは植物または動物への感染性微生物における呼吸鎖を阻害する抗菌作用を有する物質を意味する。また、エルゴステロール生合成阻害剤とは、真菌、より好ましくは植物または動物への感染性真菌におけるエルゴステロール生合成を阻害する抗菌作用を有する物質を意味する。細胞壁生合成阻害剤とは、真菌、より好ましくは植物または動物への感染性真菌における細胞壁生合成を阻害する抗菌作用を有する物質を意味する。MpkB経路阻害剤とは、真菌、より好ましくは植物または動物への感染性真菌におけるMpkBが関与するMAPキナーゼシグナル伝達経路(MpkB経路)を阻害する抗菌作用を有する物質を意味する。細胞壁構築経路阻害剤とは、細胞壁の損傷時に活性化するcell wall integrity経路を阻害する抗菌作用を有する物質を意味する。HOG経路活性化剤とは、HOG経路を活性化することで抗菌作用を有する物資を意味する。
【0026】
本発明に係るスクリーニング方法により同定された候補物質は、その抗菌作用により種々の微生物、好ましくは感染性微生物に対する抗菌剤として使用することができる。感染性微生物としては例えば、いもち病菌(Magnaporthe grisea)を挙げることができる。すなわち、本発明に係るスクリーニング方法によれば、いもち病菌(Magnaporthe grisea)等の感染性微生物に対する抗菌剤の候補物質を同定することができる。
【0027】
本発明に係るスクリーニング方法は、呼吸鎖阻害剤の指標となる遺伝子、エルゴステロール生合成阻害剤の指標となる遺伝子、細胞壁生合成阻害剤の指標となる遺伝子、MpkB経路阻害剤の指標となる遺伝子、細胞壁構築(cell wall integrity)経路阻害剤の指標となる遺伝子、及びHOG経路活性化剤の指標となる遺伝子を使用した方法(スクリーニング方法1)並びにこれら遺伝子の発現を制御するプロモータ領域及び公知のレポーター遺伝子を使用した方法(スクリーニング方法2)に大別される。
【0028】
まず、これら抗菌剤の指標となる遺伝子について説明する。
呼吸鎖阻害剤の指標となる遺伝子
呼吸鎖阻害剤の指標となる遺伝子は、従来公知の呼吸鎖阻害剤をいもち病菌(Magnaporthe grisea)に接触させた時に特異的に発現が亢進する遺伝子として見いだされた遺伝子である。ここで従来公知の呼吸鎖阻害剤とは、特に限定されないが、ジフルメトリム及びテブフェンピラド等のComplex1阻害剤、メプロニル等のComplex2阻害剤、クレソキシムメチル及びピリベンカルブ等のComplex3阻害剤、オリゴマイシン等のComplex5(F1Fo ATPase)阻害剤、フルアジナム等の電子伝達に作用してATP合成を阻害する脱共役作用剤を挙げることができる。
【0029】
呼吸鎖阻害剤の指標となる遺伝子は、下記表5に列挙したGenbankアクセッション番号によって特定される遺伝子である。下記表5における「CDS塩基配列」の欄には、Genbankアクセッション番号で特定される遺伝子のCDS領域の塩基配列を示す配列番号を記載している。また、下記表5における「BROAD ID Ver6」の欄には、Broad Instituteのマグナポルテ・グリセア(Magnaporthe grisea)ゲノムデータベースの登録番号を記載している。
【0030】
【表5】
【0031】
エルゴステロール生合成阻害剤の指標となる遺伝子
エルゴステロール生合成阻害剤の指標となる遺伝子は、従来公知のエルゴステロール生合成阻害剤をいもち病菌(Magnaporthe grisea)に接触させた時に特異的に発現が亢進する遺伝子として見いだされた遺伝子である。ここで従来公知のエルゴステロール生合成阻害剤とは、特に限定されないが、オキスポコナゾール・フマル酸塩及びフェンヘキサミドを挙げることができる。
【0032】
エルゴステロール生合成阻害剤の指標となる遺伝子は、下記表6に列挙したGenbankアクセッション番号によって特定される遺伝子である。下記表6における「CDS塩基配列」の欄には、Genbankアクセッション番号で特定される遺伝子のCDS領域の塩基配列を示す配列番号を記載している。また、下記表6における「BROAD ID Ver6」の欄には、Broad Instituteのマグナポルテ・グリセア(Magnaporthe grisea)ゲノムデータベースの登録番号を記載している。
【0033】
【表6】
【0034】
細胞壁生合成阻害剤の指標となる遺伝子
細胞壁生合成阻害剤の指標となる遺伝子は、従来公知の細胞壁生合成阻害剤をいもち病菌(Magnaporthe grisea)に接触させた時に特異的に発現が亢進する遺伝子として見いだされた遺伝子である。ここで従来公知の細胞壁生合成阻害剤とは、特に限定されないが、ミカファンギン及びカルコフロール・ホワイトを挙げることができる。
【0035】
細胞壁生合成阻害剤の指標となる遺伝子は、下記表7に列挙したGenbankアクセッション番号によって特定される遺伝子である。下記表7における「CDS塩基配列」の欄には、Genbankアクセッション番号で特定される遺伝子のCDS領域の塩基配列を示す配列番号を記載している。また、下記表7における「BROAD ID Ver6」の欄には、Broad Instituteのマグナポルテ・グリセア(Magnaporthe grisea)ゲノムデータベースの登録番号を記載している。
【0036】
【表7】
【0037】
MpkB経路阻害剤の指標となる遺伝子
MpkB経路阻害剤の指標となる遺伝子は、アスペルギルス・ニドランス(Aspergillus nidulans)の野生株と、mpkB遺伝子破壊株とにおける遺伝子の発現パターンを測定し、mpkB遺伝子破壊株において特異的に発現が亢進する遺伝子として見いだされた遺伝子である。
MpkB経路阻害剤の指標となる遺伝子は、スーパーオキシドデスムターゼ遺伝子(Genbankアクセッション番号:XM_653297、配列番号71)である。
【0038】
細胞壁構築経路阻害剤の指標となる遺伝子
細胞壁構築経路阻害剤の指標となる遺伝子は、アスペルギルス・オリゼ(Aspergillus oryzae)の野生株と、cell wall integrity経路に関与するmpkA遺伝子破壊株とにおける遺伝子の発現パターンを測定し、mpkA遺伝子破壊株において特異的に発現が亢進する遺伝子又は特異的に発現が低減する遺伝子として見いだされた遺伝子である。なお、mpkA遺伝子を破壊した株では、cell wall integrity経路が阻害されることとなる。
【0039】
細胞壁構築経路阻害剤の指標となる遺伝子は、下記表8に列挙したGenbankアクセッション番号によって特定される遺伝子である。下記表8における「CDS塩基配列」の欄には、Genbankアクセッション番号で特定される遺伝子のCDS領域の塩基配列を示す配列番号を記載している。また、下記表8における「DOGAN ID」の欄には、独立行政法人 製品評価技術基盤機構のDOGANデータベースの登録番号を記載している。
【0040】
【表8】
【0041】
なお、表8のうち、XM_001825115、XM_001820488、XM_001817008及びXM_001822481の遺伝子は、mpkA遺伝子破壊株において特異的に発現が亢進する遺伝子である。また、表8のうち、XM_001820375、XM_001727556、XM_001727176、XM_001727558、XM_001727555、XM_001820346及びXM_001827554の遺伝子は、mpkA遺伝子破壊株において特異的に発現が低減する遺伝子である。
【0042】
また、細胞壁構築経路阻害剤の指標となる遺伝子は、Fujioka et al.(2007) Eukaryotic Cell 6: 1497-1510 の論文中に記載されている、アスペルギルス・オリゼ(Aspergillus oryzae)のMpkA破壊株と野生株における転写解析のデータに基づいて選択したagsA遺伝子(Genbankアクセッション番号:XM_658397、配列番号83)及びagsB遺伝子(Genbankアクセッション番号:XM_655819、配列番号84)である。
【0043】
HOG経路活性化剤の指標となる遺伝子
HOG経路活性化剤の指標となる遺伝子は、アカパンカビ (Neurospora crassa)に関する研究でHOG経路の活性化により発現亢進することが知られるカタラーゼ遺伝子に対する、いもち病菌における相同遺伝子として見いだされた遺伝子である。また、当該相同遺伝子は、フルジオキソニル及びイプロジオン等の公知のHOG経路活性化剤に接触させた時に特異的に発現が亢進することを確認している。
HOG経路活性化剤の指標となる遺伝子cat1遺伝子(Genbankアクセッション番号:XM_365841、配列番号85)である。
【0044】
以上のように、異なる抗菌作用を有する抗菌剤の指標となる遺伝子を、Genbankアクセッション番号及び具体的な配列番号によって特定して説明したが、抗菌剤の指標となる遺伝子はCDS領域の塩基配列が上記配列番号に記載された具体的な塩基配列からなるものに限定されるものではなく、当該遺伝子に対する相同遺伝子も含む意味である。相同遺伝子とは、一般的に、共通の祖先遺伝子から進化分岐した遺伝子を意味しており、2種類の種の相同遺伝子(オルソログ(ortholog))及び同一種内で重複分岐により生じた相同遺伝子(パラログ(paralog))を含む意味である。
【0045】
より具体的には、上記配列番号に示す塩基配列に対して70%以上、好ましくは80%以上、さらに好ましくは90%以上、より好ましくは95%以上、最も好ましくは98%以上の類似度(Similarity)を有する塩基配列を含むCDS領域を有する遺伝子も、抗菌剤の指標となる遺伝子に含まれる。ここで、類似度の値は、BLAST(Basic Local Alignment Search Tool)プログラムを実装したコンピュータプログラム及び遺伝子配列情報を格納したデータベースを用いてデフォルトの設定で求められる値を意味する。
【0046】
また、より具体的には、上記配列番号に示す塩基配列に対して相補的な塩基配列を有するポリヌクレオチドにストリンジェントな条件下でハイブリダイズするcDNA又はゲノム領域から特定される遺伝子も、抗菌剤の指標となる遺伝子に含まれる。ここで、ストリンジェントな条件とは、いわゆる特異的なハイブリッドが形成され、非特異的なハイブリッドが形成されない条件をいう。例えば、45℃、6×SSC(塩化ナトリウム/クエン酸ナトリウム)でのハイブリダイゼーション、その後の50〜65℃、0.2〜1×SSC、0.1%SDSでの洗浄が挙げられ、或いはそのような条件として、65〜70℃、1×SSCでのハイブリダイゼーション、その後の65〜70℃、0.3×SSCでの洗浄を挙げることができる。ハイブリダイゼーションは、J. Sambrook et al. Molecular Cloning, A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory(1989)に記載されている方法等、従来公知の方法で行うことができる。
【0047】
スクリーニング方法1
次に、上述したスクリーニング方法1について説明する。被検物質が上述した抗菌作用を有するか評価するには、先ず、供試された被検物質を例えば感染性微生物に接触させ、被検物質の存在下における上記遺伝子の発現量と、被検物質の非存在下における発現量とを比較する。そして、公知の抗菌剤(呼吸鎖阻害剤、エルゴステロール生合成阻害剤、細胞壁生合成阻害剤、HOG経路活性化剤)を作用させた時と同様に又はmpkB遺伝子破壊株若しくはmpkA遺伝子破壊株と同様に、当該所定の遺伝子の発現量が変動している場合、当該被検物質は、上述した抗菌作用を有するものとして同定することができる。
【0048】
上述した遺伝子の発現量は、従来公知の手法により測定することができる。遺伝子の発現量を測定する手法としては、何ら限定されないが、例えばリアルタイム PCR法を適用することができる。また、マイクロアレイを用いて上述した遺伝子の発現量を測定しても良い。
【0049】
例えば、表5に示したGenbankアクセッション番号によって特定される遺伝子のうち少なくとも1以上、好ましくは5以上、より好ましくは10以上の遺伝子について、被検物質の存在下における発現量が亢進していた場合には、当該被検物質を呼吸鎖阻害剤の候補物質として同定することができる。特に、呼吸鎖阻害剤の候補物質を同定する場合、表5に示した遺伝子のうちXM_001403469で特定される遺伝子の発現量を検討することが好ましい。XM_001403469で特定される遺伝子について被検物質の存在下における発現量が亢進していた場合には、当該被検物質を呼吸鎖阻害剤の候補物質としての蓋然性が非常に高いと判断できる。
【0050】
また、例えば、表6に示したGenbankアクセッション番号によって特定される遺伝子のうち少なくとも1以上、好ましくは3以上、より好ましくは5以上の遺伝子について、被検物質の存在下における発現量が亢進していた場合には、当該被検物質をエルゴステロール生合成阻害剤の候補物質として同定することができる。特に、エルゴステロール生合成阻害剤の候補物質を同定する場合、表6に示した遺伝子のうちXM_362183で特定される遺伝子の発現量を検討することが好ましい。XM_362183で特定される遺伝子について被検物質の存在下における発現量が亢進していた場合には、当該被検物質をエルゴステロール生合成阻害剤の候補物質としての蓋然性が非常に高いと判断できる。
【0051】
さらに、例えば、表7に示したGenbankアクセッション番号によって特定される遺伝子のうち少なくとも1以上、好ましくは5以上、より好ましくは10以上、更に好ましくは20以上の遺伝子について、被検物質の存在下における発現量が亢進していた場合には、当該被検物質を細胞壁生合成阻害剤の候補物質として同定することができる。特に、細胞壁生合成阻害剤の候補物質を同定する場合、表7に示した遺伝子のうちXM_364794で特定される遺伝子及び/又はXM_368552で特定される遺伝子の発現量を検討することが好ましい。XM_364794で特定される遺伝子又はXM_368552で特定される遺伝子について被検物質の存在下における発現量が亢進していた場合には、当該被検物質を細胞壁生合成阻害剤の候補物質としての蓋然性が非常に高いと判断できる。
【0052】
さらにまた、例えば、Genbankアクセッション番号XM_653297で特定されるスーパーオキシドデスムターゼ遺伝子について被検物質の存在下における発現量が亢進していた場合には、当該被検物質をMpkB経路阻害剤の候補物質として同定することができる。
【0053】
さらにまた、例えば、表8に示したGenbankアクセッション番号によって特定される遺伝子のうち少なくとも1以上、好ましくは3以上、より好ましくは5以上の遺伝子について、被検物質の存在下における発現量が変動していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質として同定することができる。より具体的に、XM_001825115で特定される遺伝子、XM_001820488で特定される遺伝子、XM_001817008で特定される遺伝子及びXM_001822481で特定される遺伝子については、被検物質の存在下における発現量が亢進していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質として同定することができる。また、XM_001820375で特定される遺伝子、XM_001727556で特定される遺伝子、XM_001727176で特定される遺伝子、XM_001727558で特定される遺伝子、XM_001727555で特定される遺伝子、XM_001820346で特定される遺伝子及びXM_001827554で特定される遺伝子については、被検物質の存在下における発現量が減少していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質として同定することができる。特に、細胞壁構築経路阻害剤の候補物質を同定する場合、表8に示した遺伝子のうちXM_001825115で特定される遺伝子の発現量を検討することが好ましい。XM_001825115で特定される遺伝子について被検物質の存在下における発現量が亢進していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質としての蓋然性が非常に高いと判断できる。
【0054】
さらにまた、Genbankアクセッション番号XM_658397で特定されるagsA遺伝子及び/又はGenbankアクセッション番号XM_655819で特定されるagsB遺伝子について、被検物質の存在下における発現量が亢進していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質として同定することができる。
【0055】
さらにまた、Genbankアクセッション番号XM_365841で特定されるcat1遺伝子について、被検物質の存在下における発現量が亢進していた場合には、当該被検物質をHOG経路活性化剤の候補物質として同定することができる。
【0056】
以上のように、種々の被検物質について、各種の抗菌作用を遺伝子発現パターンによって判定することで、抗菌剤の候補物質をスクリーニングすることができる。ただし、本スクリーニング方法においては、ある候補物質について特定の抗菌作用の有無を判定しても良いが、ある候補物質について、呼吸鎖阻害作用、エルゴステロール生合成阻害作用、細胞壁生合成阻害作用、MpkB経路阻害作用、細胞壁構築経路阻害作用及びHOG経路活性化作用のうち何れの抗菌作用を有するのかを判定しても良い。候補物質によっては、上述した各種抗菌作用のうち複数の抗菌作用を有するものとしてスクリーニングされる場合もある。
【0057】
スクリーニング方法2
次に、上述したスクリーニング方法2について説明する。スクリーニング方法2は、スクリーニング方法1とは異なり、上述したGenbankアクセッション番号で特定される遺伝子そのものの発現量を測定するのではなく、上述したGenbankアクセッション番号で特定される遺伝子のプロモータ領域を利用する方法である。詳細には、当該プロモータ領域と当該プロモータ領域の制御下に発現可能なレポーター遺伝子とを連結した組換えDNAを使用する。この組換えDNAはベクターに組み込まれて保存・使用することができる。ベクターとしては、何ら限定されないが、導入対象の微生物において複製可能なタイプのベクターを適宜選択して使用することが好ましい。
【0058】
上述したGenbankアクセッション番号で特定される遺伝子のプロモータ領域は、当該遺伝子の発現を制御するため、当該プロモータ領域はレポーター遺伝子についても同様に発現制御する。この組換えDNAを感染性微生物に導入した形質転換細胞に対して、候補物質を接触させ、被検物質の存在下におけるレポーター遺伝子の発現量と、被検物質の非存在下における発現量とを比較する。そして、公知の抗菌剤(呼吸鎖阻害剤、エルゴステロール生合成阻害剤、細胞壁生合成阻害剤、HOG経路活性化剤)を作用させた時と同様に又はmpkB遺伝子破壊株若しくはmpkA遺伝子破壊株と同様に、当該レポーター遺伝子の発現量が変動している場合、当該被検物質は、上述した抗菌作用を有するものとして同定することができる。
【0059】
ここで、プロモータ領域とは、上述したGenbankアクセッション番号で特定される遺伝子の開始コドンから上流の領域に存在し、転写因子が結合する領域を意味する。より具体的に、プロモータ領域は、上述したGenbankアクセッション番号で特定される遺伝子の開始コドンの上流2kbの領域、好ましくは上流1.5kbの領域に含まれている。さらに具体的に、配列番号1〜84に示した84個の遺伝子の開始コドンの上流2kbの領域の塩基配列を配列番号86〜169に示す。また、配列番号85に示した遺伝子の開始コドンの上流1.5kbの領域の塩基配列を配列番号170に示す。
【0060】
本スクリーニング方法において、プロモータ領域としては、配列番号86〜170に示した塩基配列からなるポリヌクレオチドを使用することができる。また、プロモータ領域としては、配列番号86〜170に示した塩基配列に対して70%以上、好ましくは80%以上、さらに好ましくは90%以上、より好ましくは95%以上、最も好ましくは98%以上の類似度(Similarity)を有する塩基配列からなるポリヌクレオチドを使用することができる。なお、類似度(Similarity)は、上述した定義と同様である。さらに、プロモータ領域としては、配列番号86〜170に示した塩基配列に対して相補的な塩基配列を有するポリヌクレオチドにストリンジェントな条件下でハイブリダイズするポリヌクレオチドを使用することができる。なお、ストリンジェントな条件は、上述した定義と同様である。
【0061】
ここで、レポーター遺伝子としては、いわゆるレポーターアッセイに使用されるものであれば特に限定されず、如何なる遺伝子を使用しても良い。レポーター遺伝子としては、例えば、CAT遺伝子、lacZ遺伝子、ルシフェラーゼ遺伝子、β-グルクロニダーゼ遺伝子(GUS)及びGFP遺伝子等を挙げることができる。レポーター遺伝子は、上述したプロモータ領域の制御下に発現可能なように当該プロモータ領域と連結される。換言すると、プロモータ領域に転写因子が結合することにより、レポーター遺伝子の発現が誘導されるように、プロモータ領域とレポーター遺伝子とを結合する。
【0062】
レポーター遺伝子の発現レベルは、使用するレポーター遺伝子の種類に応じて、当業者に公知の方法により測定することができる。例えば、レポーター遺伝子がCAT遺伝子である場合には、該遺伝子産物によるクロラムフェニコールのアセチル化を検出することによって、レポーター遺伝子の発現レベルを測定することができる。レポーター遺伝子がlacZ遺伝子である場合には、該遺伝子発現産物の触媒作用による色素化合物の発色を検出することにより、また、ルシフェラーゼ遺伝子である場合には、該遺伝子発現産物の触媒作用による蛍光化合物の蛍光を検出することにより、また、β-グルクロニダーゼ遺伝子(GUS)である場合には、該遺伝子発現産物の触媒作用によるGlucuron(ICN社)の発光や5-ブロモ-4-クロロ-3-インドリル-β-グルクロニド(X-Gluc)の発色を検出することにより、さらに、GFP遺伝子である場合には、GFPタンパク質による蛍光を検出することにより、レポーター遺伝子の発現レベルを測定することができる。
【0063】
本スクリーニング方法においても、上述したスクリーニング方法1と同様に、例えば、表5に示したGenbankアクセッション番号によって特定される遺伝子のうち少なくとも1以上、好ましくは5以上、より好ましくは10以上の遺伝子のプロモータ領域を使用し、被検物質の存在下におけるレポーター遺伝子の発現量が亢進していた場合には、当該被検物質を呼吸鎖阻害剤の候補物質として同定することができる。特に、呼吸鎖阻害剤の候補物質を同定する場合、表5に示した遺伝子のうちXM_001403469で特定される遺伝子のプロモータ領域を用いてレポーター遺伝子の発現量を検討することが好ましい。XM_001403469で特定される遺伝子のプロモータ領域を使用した場合に、被検物質の存在下における発現量が亢進していた場合には、当該被検物質を呼吸鎖阻害剤の候補物質としての蓋然性が非常に高いと判断できる。
【0064】
また、例えば、表6に示したGenbankアクセッション番号によって特定される遺伝子のうち少なくとも1以上、好ましくは3以上、より好ましくは5以上の遺伝子のプロモータ領域を使用し、被検物質の存在下におけるレポーター遺伝子の発現量が亢進していた場合には、当該被検物質をエルゴステロール生合成阻害剤の候補物質として同定することができる。特に、エルゴステロール生合成阻害剤の候補物質を同定する場合、表6に示した遺伝子のうちXM_362183で特定される遺伝子のプロモータ領域を用いてレポーター遺伝子の発現量を検討することが好ましい。XM_362183で特定される遺伝子のプロモータ領域を使用した場合に、被検物質の存在下における発現量が亢進していた場合には、当該被検物質をエルゴステロール生合成阻害剤の候補物質としての蓋然性が非常に高いと判断できる。
【0065】
さらに、例えば、表7に示したGenbankアクセッション番号によって特定される遺伝子のうち少なくとも1以上、好ましくは5以上、より好ましくは10以上、更に好ましくは20以上の遺伝子のプロモータ領域を使用し、被検物質の存在下におけるレポーター遺伝子の発現量が亢進していた場合には、当該被検物質を細胞壁生合成阻害剤の候補物質として同定することができる。特に、細胞壁生合成阻害剤の候補物質を同定する場合、表7に示した遺伝子のうちXM_364794で特定される遺伝子のプロモータ領域及び/又はXM_368552で特定される遺伝子のプロモータ領域を用いてレポーター遺伝子の発現量を検討することが好ましい。XM_364794で特定される遺伝子のプロモータ領域又はXM_368552で特定される遺伝子のプロモータ領域を使用した場合に、被検物質の存在下における発現量が亢進していた場合には、当該被検物質を細胞壁生合成阻害剤の候補物質としての蓋然性が非常に高いと判断できる。
【0066】
さらにまた、例えば、Genbankアクセッション番号XM_653297で特定されるスーパーオキシドデスムターゼ遺伝子のプロモータ領域を使用し、被検物質の存在下におけるレポーター遺伝子の発現量が亢進していた場合には、当該被検物質をMpkB経路阻害剤の候補物質として同定することができる。
【0067】
さらにまた、例えば、表8に示したGenbankアクセッション番号によって特定される遺伝子のうち少なくとも1以上、好ましくは3以上、より好ましくは5以上の遺伝子のプロモータ領域を使用し、被検物質の存在下におけるレポーター遺伝子の発現量が変動していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質として同定することができる。より具体的に、XM_001825115で特定される遺伝子のプロモータ領域、XM_001820488で特定される遺伝子のプロモータ領域、XM_001817008で特定される遺伝子のプロモータ領域及びXM_001822481で特定される遺伝子のプロモータ領域については、被検物質の存在下におけるレポーター遺伝子の発現量が亢進していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質として同定することができる。また、XM_001820375で特定される遺伝子のプロモータ領域、XM_001727556で特定される遺伝子のプロモータ領域、XM_001727176で特定される遺伝子のプロモータ領域、XM_001727558で特定される遺伝子のプロモータ領域、XM_001727555で特定される遺伝子のプロモータ領域、XM_001820346で特定される遺伝子のプロモータ領域及びXM_001827554で特定される遺伝子のプロモータ領域については、被検物質の存在下におけるレポーター遺伝子の発現量が減少していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質として同定することができる。特に、細胞壁構築経路阻害剤の候補物質を同定する場合、表8に示した遺伝子のうちXM_001825115で特定される遺伝子のプロモータ領域を使用してレポーター遺伝子の発現量を検討することが好ましい。XM_001825115で特定される遺伝子のプロモータ領域を使用して被検物質の存在下におけるレポーター遺伝子の発現量が亢進していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質としての蓋然性が非常に高いと判断できる。
【0068】
さらにまた、Genbankアクセッション番号XM_658397で特定されるagsA遺伝子のプロモータ領域及び/又はGenbankアクセッション番号XM_655819で特定されるagsB遺伝子のプロモータ領域を使用して、被検物質の存在下におけるレポーター遺伝子の発現量が亢進していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質として同定することができる。
【0069】
さらにまた、Genbankアクセッション番号XM_365841で特定されるcat1遺伝子のプロモータ領域について、被検物質の存在下におけるレポーター遺伝子の発現量が亢進していた場合には、当該被検物質をHOG経路活性化剤の候補物質として同定することができる。
【0070】
以上のように、種々の被検物質について、各種の抗菌作用をレポーター遺伝子の発現パターンによって判定することで、抗菌剤の候補物質をスクリーニングすることができる。ただし、本スクリーニング方法においては、ある候補物質について特定の抗菌作用の有無を判定しても良いが、ある候補物質について、呼吸鎖阻害作用、エルゴステロール生合成阻害作用、細胞壁生合成阻害作用、MpkB経路阻害作用、細胞壁構築経路阻害作用及びHOG経路活性化作用のうち何れの抗菌作用を有するのかを判定しても良い。候補物質によっては、上述した各種抗菌作用のうち複数の抗菌作用を有するものとしてスクリーニングされる場合もある。
【実施例】
【0071】
以下、実施例を用いて本発明をより詳細に説明するが、本発明の技術的範囲は以下の実施例に限定されるものではない。
〔実施例1〕
呼吸鎖阻害剤検出マーカーの選抜方法
呼吸鎖にその作用を示す薬剤を検出することのできるマーカー遺伝子の選抜方法を以下に示す。
【0072】
呼吸鎖阻害剤暴露状態でのマイクロアレイによる発現解析
(1)トータルRNA抽出
トータルRNA抽出に用いた菌体は、100mlのYG液体培地を入れた300ml容三角フラスコにマグナポルテ・グリセア野生株Guy11株の分生子を約2×106個分生子になるように接種し、26℃、48時間緩やかに振とう培養後、呼吸鎖Complex1阻害剤としてジフルメトリム及びテブフェンピラド、Complex2阻害剤としてメプロニル、Complex3阻害剤としてクレソキシムメチル及びピリベンカルブ、Complex5(F1Fo ATPase)阻害剤としてオリゴマイシン、電子伝達に作用してATP合成を阻害する脱共役作用剤としてフルアジナムを0.01〜30ppmの濃度添加し、さらに26℃で2時間培養した。本薬剤処理を行った菌体及び無処理菌体は、ミラクロス(CALBIOCHEM社製)で集菌した。この回収した菌を乳鉢中で液体窒素を注ぎながらパウダー状になるまで破砕した。パウダー状の菌体はRNA Plant Mini Kit(キアゲン社製)を用いて、添付マニュアルに従いトータルRNA抽出を行った。
【0073】
(2)マイクロアレイ
マイクロアレイに用いるDNAチップはマグナポルテ・グリセアの10,000遺伝子から設計した60merのオリゴプローブをインクジェット方式でスポットした独自のマイクロアレイを使用し、抽出した薬剤処理区をtestサンプルに、薬剤無処理区を比較対象にCyanine3及びCyanine5を用いた二色法により行った。一回の実験区ごとにCyanine3とCyanine5のカラースワップを行う事により計2回の反復実験を行った。
【0074】
(3)RNAの標識(Cyanine5、Cyanine3)
RNAのCyanine3及びCyanine5標識はmRNAを用いて行い、トータルRNAからのmRNAの精製はOligotex-dT30<super> mRNA purification Kit(タカラバイオ社製)を用い、本キットに添付のマニュアルに従った。mRNAからのcDNAの作製及び標識はCyScribe cDNA Post Labelling Kit(GE helthcare)を用い、本キットに添付のマニュアルに従い行った。
【0075】
(4)ハイブリダイゼーション
Cyanine3及びCyanine5標識済みcDNA混合サンプルに、表9に示したハイブリダイゼーションBufferを60μl加え溶解後、95℃で5分熱変性を行った。その後30分間室温で保持した後、DNAチップ上に滴下し、カバーガラスをのせた。カバーガラスをのせたDNAチップは温水上に浮かべたhumidity chamber上に置き42℃、一晩インキュベーションすることによりハイブリダイゼーションを行った。
【0076】
【表9】
【0077】
(5)ポストハイブリダイゼーション
42℃で一晩インキュベーション後、DNAチップは表10に示したWash buffer1中でカバーガラスをはずした後、さらに新しいWash Buffer1で15分、次いで表11に示したWash Buffer2で5分、表12に示したWash Buffer3で5分の順に振とうすることにより洗浄を行った。その後、さらにdH2O中で2分間洗浄後800×g、2分の遠心により水分を飛ばしGenePix 4000B(Axon instrument)を用いてスポットのスキャンを行った。
【0078】
【表10】
【0079】
【表11】
【0080】
【表12】
【0081】
マイクロアレイの結果から抽出された呼吸鎖阻害剤処理により共通して発現する遺伝子のうち、呼吸鎖阻害剤及びその他抗菌剤の薬物代謝に関わる遺伝子を除いた遺伝子を呼吸鎖阻害剤の検出マーカー候補とした。以下の表13にその上位10遺伝子のマイクロアレイ解析結果を表示する。値は無処理区と比較した場合の相対値で示し、発現比1.5倍以上の値を網掛けで表示した。
【0082】
【表13】
【0083】
〔実施例2〕
呼吸鎖阻害剤検出マーカー遺伝子の検証
表13に示した遺伝子が呼吸鎖阻害剤を処理したときに、特異的に発現変化があり検出マーカーとして利用可能か否かをMGG_12936シアン耐性呼吸酵素遺伝子(AOX)及びMGG_04999NADH-デヒドロゲナーゼ遺伝子を例に検証した。
【0084】
(1)トータルRNA抽出
トータルRNA抽出に用いた菌体は、100mlのYG液体培地を入れた300ml容三角フラスコにマグナポルテ・グリセア野生株Guy11株の分生子を約2×106個分生子になるように接種し26℃、48時間緩やかに振とう培養。その後、指定の薬剤を各濃度添加し、さらに26℃で30分、2時間及び6時間培養した。薬剤処理に用いた呼吸鎖阻害薬剤及び処理濃度は呼吸鎖Complex1阻害剤としてジフルメトリム、Complex2阻害剤としてメプロニル、Complex3阻害剤としてクレソキシムメチル、Comlex5(F1Fo ATPase)阻害剤としてオリゴマイシン、電子伝達に作用してATP合成を阻害する脱共役作用剤としてフルアジナムをそれぞれ、1ppm、30ppm、20ppm、0.5ppm、0.01ppmとした。その他、対照薬剤として、フラバノン系化合物サクラネチン10ppm、MAPキナーゼシグナリングに作用すると考えられるフルジオキソニル20ppm、プロシミドン10ppm、イプロジオン30ppm、エルゴステロール生合成阻害剤オキスポコナゾール・フマル酸塩1ppmを用いた。本薬剤処理により得た菌体及び無処理菌体は、ミラクロス(CALBIOCHEM社製)で集菌した。回収した菌を乳鉢中で液体窒素を注ぎながらパウダー状になるまで破砕した。パウダー状の菌体はRNA Plant Mini Kit(キアゲン社製)を用いて、添付マニュアルに従いトータルRNA抽出を行った。
【0085】
(2)cDNAの合成
cDNAの合成には、トータルRNAを用いた。260 nmにおける吸光度からの計算値で5μg量のトータルRNAをRNase freeの1.5mlチューブに移し、さらにDEPC処理水を加え100μlとした。これにRNase free DNase Iを5unit加え、37℃で1時間反応させ混在するゲノムDNAの分解を行った。その後、ExScript RT reagent Kit(タカラバイオ社製)を用いて添付マニュアルに従って逆転写反応を行いcDNAを得た。
【0086】
(3)定量RT-PCR
合成したcDNAの蛍光色素によるラベリング反応には、DyNAmo SYBR Green qPCR Kit (Finnzymes社製)を用いた。スキャナー解析はDNA Engine OPTICON2(MJ Research社製)を、データ解析には解析ソフトOpticon Monitor2 ver.2.02(MJ Research社製)を用いた。反応操作は、DyNAmo SYBR Green qPCR Kit添付マニュアルにしたがった。反応条件は95℃、10分間のヒートショック後、熱変性95℃、10秒、アニーリング60℃、30秒、伸長反応72℃、30秒、プレートリードの反応を40 サイクル行い、60〜95℃のグラジェント(0.2℃おきに1秒間hold)でDissociation Curveの作成を行った。PCRは各cDNA、各標的遺伝子につき三連で行った。
【0087】
呼吸鎖阻害剤処理cDNAサンプルを用いてシアン耐性呼吸酵素遺伝子(AOX)及びNADH-デヒドロゲナーゼ遺伝子の発現を経時的に調べた。またコントロールとしてActinタンパク質をコードしていると推定される遺伝子の転写量を調べた。それぞれ以下の特異的プライマーを用いた。
Actin-F:ATGCCATCGGAAAGACAGAC(配列番号171)
Actin-R:CAGGAGTCGATCTCCAAAGC(配列番号172)
AOX_RTF:TTGTCGCATACCTCATCAGC(配列番号173)
AOX_RTR:CAATTTCTGGCACCTGGAAC(配列番号174)
NADH_RTF:AATCACCAAGGATGGACGAC(配列番号175)
NADH_RTR:CCTAGGTATGCCATGGTTCC(配列番号176)
【0088】
PCR終了後、専用の解析ソフトOpticon Monitor2 ver.2.02(MJ Research社)を用いて解析を行った。Quantitation画面上で各遺伝子のPCR産物が対数的に増幅し始めるサイクル数(CT)を決定した。この値をもとに各cDNAの標的遺伝子の相対発現量を以下の計算方法から求めた。
a) ΔCT値の算出(各cDNAの標的遺伝子とγ-actinとの差)
ΔCT = (y-x)
ここでxはCT (γ-actin)であり、yはCT(Target gene)である
b) ΔCT値の算出(あるcDNAのΔCTと基準とするcDNAのΔCTとの差)
ΔCT =ΔCT (C) -ΔCT (S)
ここで、CはComparative cDNAであり、SはStandard cDNAである
c) 相対発現量の算出
2-(ΔCT)
【0089】
RT-PCRの結果をシアン耐性呼吸酵素遺伝子(AOX)については図1の上段に、NADH-デヒドロゲナーゼ遺伝子については図1の下段にactin当たりの発現量をグラフ化したものを示した。野生株を呼吸鎖阻害剤に曝すと、シアン耐性呼吸酵素遺伝子(AOX)及びNADH-デヒドロゲナーゼ遺伝子の転写量は増加していた。一方、呼吸鎖への作用機作を有しない化合物に関しては、それぞれの遺伝子発現の顕著な増加は認められなかった。このことにより、今回例として設定したシアン耐性呼吸酵素遺伝子(AOX)及びNADH-デヒドロゲナーゼ遺伝子は呼吸鎖に作用を及ぼす薬剤特異的に発現が増加していると考えられる。よって、例えば、シアン耐性呼吸酵素遺伝子 (AOX)及びNADH-デヒドロゲナーゼ遺伝子の5'-UTR(5'上流領域のプロモータ領域)はレポーター遺伝子と機能的に結合させることにより、呼吸鎖阻害剤のスクリーニング系として利用できる可能性が確認された。
【0090】
〔実施例3〕
呼吸鎖阻害剤検出レポーター
実施例2で呼吸鎖阻害剤を暴露した時に特異的な遺伝子の発現増を示すシアン耐性呼吸酵素遺伝子(AOX)を例にして、本遺伝子の5'-UTR(5'上流領域のプロモータ)をレポーター遺伝子と機能的に結合させることによって転写量の呼吸鎖特異的な増加を可視化するシステムを構築した例を示す。
【0091】
(1)シアン耐性呼吸酵素遺伝子(AOX) 5'領域のクローニング
Broad Institute のマグナポルテ・グリセア(Magnaporthe grisea)ゲノムデータベース(http://www.broad.mit.edu/annotation/genome/magnaporthe_grisea/MultiHome.html)からAOX 遺伝子の5'領域の塩基配列情報を取得し、その配列情報を参考に一組のオリゴヌクレオチドを設計した(5'-CCAAGCTTCCTTCCTCACTA-3'(配列番号177)、5'-CCAAGCTTAAGACGGTACGTTCAGTGT-3'(配列番号178))。マグナポルテ・グリセアGuy11株のゲノムDNAをテンプレートとしてPCR反応を行った。増幅反応は、94℃、3分間鋳型DNAを変性し、94℃、30秒間、55℃、30秒間、72℃、2分間保持するサイクルを35サイクルおこなった後、72℃、5分間で完全伸長させ、4℃で保持した。PCR用装置は、PCR Thermal Cycler PERSONAL (タカラバイオ社製)を用いた。このPCRによる増幅断片をアガロース電気泳動にて確認を行ったところ約889塩基対の増幅が見られた。この増幅断片をアガロースゲル電気泳動に供したゲル中よりGENECLEAN IIIキット(Qbiogene社製)を用いてDNAを抽出し、これを挿入DNA断片とした。この挿入DNA断片をpGEM-T Easy Vector System(Promega社製)を用いてpGEM-T Easy Vectorに連結させ、連結DNA溶液を得た。連結DNA溶液10μlを氷中でよく冷却した後、氷上解凍したコンピテントセルJM109を100μl加え穏やかに撹拌し、氷中で20分間、続いて42℃で45秒ヒートショック処理を行った。これに、400μlのLB液体培地を加え、37℃で1時間培養後、100μg/mlのアンピシリンを添加したLB平板培地にまき、37℃で一晩培養した。目的のプラスミドDNAを用いて形質転換した大腸菌の単一のコロニーを3mlの100μg/mlのアンピシリンを添加したLB液体培地に植菌し、37℃で一晩振盪培養した。1.5mlの培養液を1.5mlのエッペンドルフチューブに移して15,000×gで1分間遠心分離し、沈殿を100μlの氷冷したTEG(25mM Tris-HCl、10mM EDTA、50mM Glucose、pH8.0)で懸濁し、これに200μlの0.2N NaOH-1%SDSを加えて穏やかに撹拌した後、150μlの3M NaOAc(pH5.2)を加えて混合した。これを15,000×g、4℃で5分間遠心分離し上清を回収し450μlのフェノール・クロロホルム・イソアミルアルコール(25:24:1)を加えて激しく撹拌した後、15,000×g、室温で5分間遠心分離し上層を回収した。この溶液に-20℃で氷冷した900μlのエタノールを加えて-20℃で10分間放置後、4℃にて15,000×gで5分間遠心分離した。沈殿を500μlの70%エタノールでリンスしたのち、乾燥させ、最後にRNase(100μg/ml)を含むTE(10mM Tris-HCl、1mM EDTA、pH8.0)50μlに溶解した。得られたプラスミドをHindIIIで切断後、アガロース電気泳動により挿入断片の存在を確認し、このプラスミド中の挿入DNA断片を添付のプロトコールに従い、ABI PRISMTM 377 DNA sequencing system (PE Biosystem社製)にて解析した。889塩基対からなるシアン耐性呼吸酵素遺伝子(AOX)5'上流領域のプロモータの塩基配列を配列番号179に示す。また、本ベクターをpGEM-PrAOXとする。
【0092】
(2)EGFPベクターの作成
pCAMBIA-0380バイナリーベクター(CAMBIAより購入)を骨格にし、薬剤選択マーカーとしてハイグロマイシンB耐性遺伝子Hphを融合することにより作成した糸状菌形質転換ベクターpCH11(図2)をHindIII及びBglIIで消化し、そこにEGFP-LacI-TagdA (p3'SS d EGFPと大腸菌LacI:Lactose operon transcriptional repressor Lac1(E. coli EU337980, 4-1068bp)及びAspergillus oryzae Α-グルコシダーゼ遺伝子agdA ターミネーター(Aspergillus oryzae、Supercontig 6: 3289259-3289838 +)融合体)のHindIII-BglII断片を組み合すことによりEGFPベクター、pCH11-EGFP-LacIを作成した(図3)。
【0093】
(3)AOX-EGFP-LacIレポーター(レポーター遺伝子ベクター)の作製
pCH11-EGFP-LacI(図2)プラスミドをHindIIIで消化し、そこにpGEM-PrAOXをHindIII処理することにより得たシアン耐性呼吸酵素遺伝子(AOX)5'領域を定法により融合した。本レポーターアッセイプラスミドであるAOX-EGFP-LacIレポーターベクターをpCH11-PrAOX-EGFP-LacIと命名した。
【0094】
(4) pCH11-PrAOX-EGFP-LacIプラスミドを用いたマグナポルテ・グリセアの形質転換
マグナポルテ・グリセアの形質転換はアグロバクテリウム法(Gento Tsuji,Satoshi Fujii, Naoki Fujihara, Chika Hirose,Seiji Tsuge, Tomonori Shiraishi and Yasuyuki Kubo(2003):Agrobacterium tumefaciens-mediated transformation for random insertional mutagenesis in Colletorichum lagenarium. Journal of General Plant Pathology 69:230-239)を改良した方法を用いた。得られた菌株へのベクター挿入の確認は、PCR、シークエンス及びサザン解析によって行った。
【0095】
(5)レポーター作動の確認
レポーター作動の確認は、呼吸鎖阻害剤であるジフルメトリム、クレソキシムメチル、オリゴマイシン及び比較薬剤としてオキスポコナゾール、フェンヘキサミド、カルプロパミド、ベノミルで菌体を処理した時のEGFPの蛍光を観察することによって行った。マグナポルテ・グリセアPAOX-NLS株形質転換株の分生子を2×106個分生子/100mlになるように、100mlのYG液体培地を入れた300ml容坂口フラスコに接種し次の条件で培養した。
(条件1) 26℃、24時間培養
(条件2) 26℃、24時間培養した後、上記薬剤を添加し、さらに26℃で6時間培養。
その後、各処理菌株の蛍光を蛍光顕微鏡を用いて観察した。
【0096】
(6)EGFP蛍光観察
スライドガラスに培養後の菌糸体をのせGFPの蛍光観察を行った。各種薬剤添加6時間後、呼吸鎖阻害剤(ジフルメトリム、クレソキシムメチル、オリゴマイシン)処理においてのみ、核に局在した強いEGFP-LacI蛍光が観察され、無処理区及び呼吸鎖阻害剤以外の化合物処理ではEGFP-LacI蛍光はほとんど観察されなかった(図4)。以上の結果から、pAOX-EGFP-LacIレポーターは呼吸鎖阻害剤の特異的スクリーニングに利用できると言える。
【0097】
〔実施例4〕
エルゴステロール生合成阻害剤検出マーカーの選抜方法
エルゴステロールの生合成にその作用を示す薬剤を検出することのできるマーカー遺伝子の選抜方法は、呼吸鎖阻害剤の代わりにエルゴステロール生合成阻害剤であるオキスポコナゾール・フマル酸塩を1及び5ppm、フェンヘキサミドを0.5ppmの濃度で使用した以外は実施例1と同様に行った。
【0098】
マイクロアレイの結果から抽出されたエルゴステロール生合成阻害剤で共通して発現する遺伝子のうち、エルゴステロール生合成阻害剤及びその他抗菌剤の薬物代謝に関わる遺伝子を除いた遺伝子をエルゴステロール生合成阻害剤の検出マーカー候補とした。以下の表14にその上位5遺伝子のマイクロアレイ解析の結果を表示する。値は無処理区と比較した場合の相対値で示し、発現比1.5倍以上の値を網掛けで表示した。
【0099】
【表14】
【0100】
〔実施例5〕
エルゴステロール生合成阻害剤検出マーカー遺伝子の検証
表14に示した遺伝子がエルゴステロール生合成阻害剤を処理したときに、特異的に発現変化があり検出マーカーとして利用可能か否か、Cytochrome P450ステロール14αジメチラーゼ(MGG_04628)及びステロール24Cメチルトランスフェラーゼ(MGG_10860)を例に検証した。
【0101】
本実施例では、Cytochrome P450ステロール14αジメチラーゼ(MGG_04628)及びステロール24Cメチルトランスフェラーゼ(MGG_10860)を特に検出するため、以下の特異的プライマーを使用した以外は実施例2と同様にして遺伝子の転写量を調べた。
SteCyp51-F:CCACATGATGATCACGCTTC(配列番号180)
SteCyp51-R:GTCTCCTTGACCACGTTTGC(配列番号181)
24C-F:AACATGCAGACAGCCAAGAC(配列番号182)
24C-R:ATTCGAAATGTTCCCAGCAC(配列番号183)
【0102】
RT-PCRの結果をCytochrome P450ステロール14αジメチラーゼ(MGG_04628)については図5の上段に、ステロール24Cメチルトランスフェラーゼ(MGG_10860)については図5の下段にactin 当たりの発現量をグラフ化したものを示した。野生株を各種薬剤に曝すと、Cytochrome P450ステロール14αジメチラーゼ(MGG_04628)及びステロール24Cメチルトランスフェラーゼ(MGG_10860)の転写量はエルゴステロール生合成阻害剤オキスポコナゾール添加後増加していた。このことにより、今回例として設定したCytochromeP450ステロール14αジメチラーゼ(MGG_04628)及びステロール24Cメチルトランスフェラーゼ(MGG_10860)はエルゴステロール生合成に作用を及ぼす薬剤処理下で発現が増加していると考えられる。よって、例えば、Cytochrome P450ステロール14αジメチラーゼ(MGG_04628)及びステロール24Cメチルトランスフェラーゼ(MGG_10860)の5'-UTR(5'上流領域のプロモーター)は、レポーター遺伝子と機能的に結合させることにより、エルゴステロール生合成阻害剤のスクリーニング系として利用できる可能性が確認された。
【0103】
〔実施例6〕
エルゴステロール生合成阻害剤検出レポーター
実施例5でエルゴステロール生合成阻害剤を暴露した時に、特異的な遺伝子の発現増を示すCytochromeP450ステロール14αジメチラーゼ(MGG_04628)を例にして、本遺伝子の5'-UTR(5'上流領域のプロモーター)をレポーター遺伝子と機能的に結合させることによって転写量のエルゴステロール生合成阻害剤特異的な増加を可視化するシステムを構築した例を示す。
【0104】
Broad Instituteのマグナポルテ・グリセア(Magnaporthe grisea)ゲノムデータベース (http://www.broad.mit.edu/annotation/genome/magnaporthe_grisea/MultiHome.html)からCytochrome P450ステロール14αジメチラーゼ(MGG_04628)の5'領域の塩基配列情報を取得し、その配列情報を参考に一組のオリゴヌクレオチドを設計した(5'-CCAAGCTTATGTGAGCCATCTTCGGATT-3'(配列番号184)、5'-CCAAGCTTGACGATCGGGTAAGGAGACT-3'(配列番号185))。マグナポルテ・グリセアGuy11株のゲノムDNAをテンプレートとしてPCR反応を行った。増幅反応は、94℃、3分間鋳型DNAを変性し、94℃、30秒間、55℃、30秒間、72℃、2分間保持するサイクルを35サイクルおこなった後、72℃、5分間で完全伸長させ、4℃で保持した。PCR用装置はPCR Thermal Cycler PERSONAL(タカラバイオ社製)を用いた。このPCRによる増幅断片をアガロース電気泳動にて確認を行ったところ約1,405塩基対の増幅が見られた。
【0105】
その後、実施例3と同様にしてプロモータ領域の塩基配列を解析した結果を配列番号186に示す。また、本ベクターをpGEM-PrCyp51とする。
【0106】
また、実施例3と同様にして、Cyp51-EGFP-LacIレポーターを作製し、レポーター作動を確認した。その結果を図6に示した。各種薬剤添加6時間後、オキスポコナゾール・フマル酸塩の処理においてのみ、核に局在した強いEGFP-LacI蛍光が観察され、それ以外の化合物処理ではEGFP-LacI蛍光はほとんど観察されなかった。以上の結果から、pCyp51-EGFP-LacIレポーターシステムはエルゴステロール生合成阻害剤のスクリーニングに利用できると言える。
【0107】
〔実施例7〕
細胞壁生合成阻害剤の検出マーカーの選抜方法
細胞壁の生合成にその作用を示す薬剤を検出することのできるマーカー遺伝子の選抜方法は、呼吸鎖阻害剤の代わりに細胞壁生合成阻害剤である1μgミカファンギン(アステラス製薬)及び1mgカルコフロール・ホワイト(シグマ・アルドリッチ)を使用した以外は実施例1と同様に行った。
【0108】
マイクロアレイの結果から抽出された細胞壁生合成阻害剤により共通に発現が誘導される、もしくは抑制される遺伝子のうち、細胞壁生合成阻害剤の薬物代謝に関わる遺伝子を除いた遺伝子を細胞壁生合成阻害剤の検出マーカー候補とした。以下の表15にそのマイクロアレイ結果の一部を表示する。値は無処理区と比較した場合の相対値で示した。
【0109】
【表15】
【0110】
〔実施例8〕
細胞壁生合成阻害剤検出レポーター
表15に示した遺伝子が細胞壁生合成阻害剤を処理したときに、特異的に発現変化があり検出マーカーとして利用可能か否か検証した。本実施例では、以下の特異的プライマーを使用した以外は実施例2と同様にして遺伝子の転写量を調べた。
MGG_02004-F:GGACAAAATGACAGTCCA(配列番号187)
MGG_02004-R:CCAAGTTTTCTTTCGGTTTC(配列番号188)
MGG_00692-F:TACAGACATGCCGCCGGGTGGACAA(配列番号189)
MGG_00692-R:ACGCTTGAGGTCCATCTGACCACTGCT(配列番号190)
MGG_04943-F:CCTTCTCGATCGCATGCT(配列番号191)
MGG_04943-R:ATCTGACCGACGTACTCTTG(配列番号192)
MGG_09639-F:TGTTTTCGTCGCAGTTCGTC(配列番号193)
MGG_09639-R:GCTCCTTTCTAAAGACCTTGGA(配列番号194)
MGG_05133-F:GTTGTTTCTGAGACCGGCCTGGTCTAC(配列番号195)
MGG_05133-R:CCATCGCTGATGGGTCCATGCCTACA(配列番号196)
MGG_02773-F:GATCATGGAACGGCAAAG(配列番号197)
MGG_02773-R:GTACCCCATTTCACCAAAC(配列番号198)
【0111】
RT-PCRの結果を図7にactin当たりの発現量をグラフ化したものを示した。野生株を細胞壁ストレス剤に曝すと、マイクロアレイの結果と同様に、4遺伝子(MGG_02004、MGG_00692、MGG_04943及びMGG_09639)の転写量は細胞壁生合成阻害剤添加後増加し、2遺伝子(MGG_05133及びMGG_02773)の転写量は減少していた。このことにより、今回例として設定した上記6遺伝子は細胞壁生合成に作用を及ぼす薬剤特異的に発現が変動していると考えられる。よって、例えば、これらの5'-UTR(5'上流領域のプロモータ)はレポーター遺伝子と機能的に結合させることによって、本発明のスクリーニング方法においてレポーター遺伝子の転写量の変化を検出することにより、細胞壁生合成阻害剤のスクリーニング系として利用できる可能性が確認された。
【0112】
〔実施例9〕
細胞壁生合成阻害剤のスクリーニングに用いるレポーターアッセイ株の作出
実施例8で細胞壁生合成阻害剤を曝露した時に、特異的な遺伝子の発現増を示す2遺伝子(MGG_00692及びMGG_09639)を例にして、これら遺伝子の5'-UTR(5'上流領域のプロモータ)をレポーター遺伝子と機能的に結合させることによって転写量の細胞壁生合成阻害剤特異的な増加を可視化するシステムを構築した例を示す。
【0113】
(1)遺伝子(MGG_00692) 5'領域のクローニング
Broad Instituteのマグナポルテ・グリセア(Magnaporthe grisea)ゲノムデータベース(http://www.broad.mit.edu/annotation/genome/magnaporthe_grisea/MultiHome.html)から遺伝子(MGG_00692)の5'領域の塩基配列情報を取得し、その配列情報を参考に一組のオリゴヌクレオチドを設計した(5'-CTTAGGCAACCTTGACGGCC-3'(配列番号199)、5'-GTCGTGTCCTTGGTTCATGG-3'(配列番号200))。マグナポルテ・グリセアGuy11株のゲノムDNAをテンプレートとしてPCR反応を行った。増幅反応は、94℃、3分間鋳型DNAを変性し、94℃、30 秒間、55℃、30秒間、72℃、2分間保持するサイクルを35サイクルおこなった後、72℃、5分間で完全伸長させ、4℃で保持した。PCR用装置はPCR Thermal Cycler PERSONAL(タカラバイオ社製)を用いた。このPCRによる増幅断片をアガロース電気泳動にて確認を行ったところ約1,000塩基対の増幅が見られた。
【0114】
その後、実施例3と同様にしてプロモータ領域の塩基配列を解析した結果を配列番号201に示す。また、本ベクターをpGEM-MSTU1とする。
【0115】
(2)遺伝子(MGG_09639)5'領域のクローニング
Broad Instituteのマグナポルテ・グリセア(Magnaporthe grisea)ゲノムデータベース (http://www.broad.mit.edu/annotation/genome/magnaporthe_grisea/MultiHome.html)から遺伝子(MGG_09639)の5'領域の塩基配列情報を取得し、その配列情報を参考に一組のオリゴヌクレオチドを設計した(5'-GTCCCGACAGCCAGTGTGCA-3'(配列番号202)、5'-GTTTTTTTACGATACCACCATAAAGTG-3'(配列番号203))。マグナポルテ・グリセアGuy11株のゲノムDNAをテンプレートとしてPCR反応を行った。増幅反応は、94℃、3分間鋳型DNAを変性し、94℃、30 秒間、55℃、30秒間、72℃、2分間保持するサイクルを35サイクルおこなった後、72℃、5分間で完全伸長させ、4℃で保持した。PCR用装置は PCR Thermal Cycler PERSONAL(タカラバイオ社製)を用いた。このPCRによる増幅断片をアガロース電気泳動にて確認を行ったところ約1,000塩基対の増幅が見られた。
【0116】
その後、実施例3と同様にしてプロモータ領域の塩基配列を解析した結果を配列番号204に示す。また、本ベクターをpGEM-AGS1とする。
【0117】
(3)EGFPベクターの作成
Aspergillus nidulans用シャトルベクター pAUR316(タカラバイオ社製)の自律複製AMA配列を除いたpNA316のBgl IIサイトに、EGFP-LacI-NLS-TagdA (p3'SS dEGFPと大腸菌LacI: Lactose operon transcriptional repressor Lac1(E. coli EU337980, 4-1068bp)及びSV40の核局在配列、Aspergillus oryzae Α-グルコシダーゼ遺伝子agdAターミネーター(Aspergillus oryzae、Supercontig 6: 3289259-3289838 +)融合体)を組み合わせたEGFPベクターpNA(N)EGFPを構築した(図8)。
【0118】
(4)pMSTU1-EGFPレポーター(レポーター遺伝子ベクター)の作製
pNA(N)EGFP(図8)プラスミドをNotIで消化し、そこにpGEM-MSTU1をNotI処理することにより得た 遺伝子MGG_00692の5'領域を定法により融合した。本レポーターアッセイプラスミドはpMSTU1-EGFPと命名した。
【0119】
(5)pAGS1-EGFPレポーター(レポーター遺伝子ベクター)の作製
pNA(N)EGFP(図8)プラスミドをNotIで消化し、そこにpGEM-AGS1をNotI処理することにより得た 遺伝子MGG_09639の5'領域を定法により融合した。本レポーターアッセイプラスミドはpAGS1-EGFPと命名した。
【0120】
(6)pMSTU1-EGFP及びpAGS1-EGFPを用いたマグナポルテ・グリセアの形質転換
マグナポルテ・グリセアの形質転換はPEG-プロトプラスト法(Leung, H., Lehtinen, U., Karjalainen, R., Skinner, D., Tooley, P., Leong, S., Ellingboe, A., 1990. Transformation of the rice blast fungus Magnaporthe grisea to hygromycin B resistance. Curr. Genet. 17, 409-411.)によって行った。得られた菌株へのベクター挿入の確認は、PCR、シークエンス及びサザン解析によって行った。
【0121】
(7)レポーター作動の確認
レポーター作動の確認は、実施例3と同様に行った。スライドガラスに培養後の菌糸体をのせGFPの蛍光観察を行った結果を図9に示す。細胞壁生合成阻害剤添加3時間後の処理において核に局在した強い緑色蛍光が観察され、無処理区では緑色蛍光はほとんど観察されなかった。以上の結果から、pMSTU1-EGFPレポーター及びpAGS1-EGFPレポーターは細胞壁生合成阻害剤の特異的スクリーニングに利用できる。
【0122】
〔実施例10〕
Aspergillus nidulans mpkB遺伝子破壊株の造成
(1)遺伝子欠失破壊用DNA断片の作製
アスペルギルス・オリゼ(Aspergillus oryzae)のargB遺伝子を欠失破壊株の選択用マーカーとして選択し、argB遺伝子を含むpAORBプラスミドから以下のように設計したプライマーを用いてargB遺伝子の断片をPCR増幅した(5'-GAATTCCATCCGCTGTGGCCGACTCA-3' (配列番号205)、5'-GGTACCGACGGAGTAACTGGAAAGATACGA-3' (配列番号206))。増幅にはTAKARAのPrimeStar HS DNA polymerase を用い、増幅反応は、95℃、2分間鋳型DNAを変性し、98℃、10秒間、55℃、5秒間、72℃、2分30秒間保持するサイクルを30サイクルおこなった後、72℃、5分間で完全伸長させ、4℃で保持した。このPCRによる増幅断片をアガロースゲル電気泳動にて確認を行ったところ約2700塩基対の増幅が見られた。
【0123】
次に、Broad Instituteのアスペルギルス属ゲノムデータベース(http://www.broad.mit.edu/annotation/genome/aspergillus_group/MultiHome.html)からアスペルギルス・ニドランス(Aspergillus nidulans)のmpkB遺伝子を含む上下流域の塩基配列を参考にプライマーを設計し、mpkB遺伝子の開始コドンから5'側上流領域(5'-UTR)と終止コドンから3'側下流領域(3'-UTR)の各1kbのDNA領域をPCRで増幅した。増幅にはタカラバイオ社製のPrimeStar HS DNA polymeraseを用い、増幅反応は、95℃、2分間鋳型DNAを変性し、98℃、10秒間、55℃、5秒間、72℃、2分間保持するサイクルを30サイクルおこなった後、72℃、5分間で完全伸長させ、4℃で保持した。このPCRによる増幅断片をアガロースゲル電気泳動にて確認を行ったところ約1,000塩基対の増幅が見られた。5'-UTR、前述のargB遺伝子断片、3'-UTRをfusion PCRにより連結する訳であるが、argB遺伝子断片との連結部になる位置にアニールするプライマーのテール部には、argB遺伝子断片の末端の配列を35 base導入した。各プライマーの配列を表16に記した。
【0124】
【表16】
【0125】
5'-UTR、argB遺伝子、3'-UTRの各断片をアガロースゲル電気泳動に供したゲル中よりillustra GFX PCR DNA and Gel Band Purification Kit(GE healthcare社製)を用いてDNAを抽出した。これら3断片を等量ずつ混合したDNA溶液をPCRの鋳型とし、fusion PCRを行った。fusion PCRは、両端に位置するANmpkB-5'UTR-FとANmpkB-3'UTR-Rのプライマーを供し、PrimeStar HS DNA polymerase(タカラバイオ社製)を用いて以下に記す増幅反応をおこなった。すなわち、94℃、2分間鋳型DNAを変性し、94℃、30秒間、58℃、45秒間、72℃、5分間保持するサイクルを30サイクルおこなった後、72℃、10分間で完全伸長させ、4℃で保持した。このPCRによる増幅断片をアガロースゲル電気泳動にて確認を行ったところ、3つの断片の合計サイズと同じ約4,700塩基対の増幅が見られた。この断片をアガロースゲル電気泳動に供したゲル中よりillustra GFX PCR DNA and Gel Band Purification Kit(GE healthcare社製)を用いてDNAを抽出し、mpkB遺伝子欠失破壊用DNA断片とした。
【0126】
(2)形質転換
mpkB遺伝子欠失破壊のための形質転換はプロトプラスト-PEG法を改良した方法を用い、形質転換するDNA断片は、上記で作製したmpkB遺伝子欠失破壊用DNA断片を用いた。アスペルギルス・ニドランスABPU1 ΔligD::ptrA(ビオチン(biA1)、アルギニン(argB2)、ウリジン(pyrG89)、ピリドキシン(pyroA4)要求性株(東北大学大学院農学研究科・藤岡智則博士より供与))の分生子懸濁液をYPD培地に植菌し、37℃、20時間振盪培養した。17G1滅菌済ガラスフィルターを用いて集菌後、菌体を50 ml容の遠心チューブに移し、滅菌水で洗浄した。その後、菌体を30 mlのプロトプラスト化溶液(0.8M NaCl、10mM NaH2PO4、10mg/ml Lysing enzyme (Sigma Chemical社製)、5mg/ml Cellulase Onozuka R-10 (Yakult Pharmaceutical Ind.社製)、2.5mg/ml Yatalase (タカラバイオ社製)を加え懸濁し、30℃、90rpm、3時間振盪しプロトプラスト化反応を行った。滅菌したMIRACLOTH(CALBIOCHEM社製)にて濾過し、濾液中のプロトプラストを3,000×g、4℃、5分間遠心分離して沈澱として得た。0.8M NaClにて1回プロトプラストを洗浄し、3,000×g、4℃、5分間遠心分離して沈澱させた。このプロトプラストをSolution 1(0.8M NaCl、10mM CaCl2、10mM Tris-HCl、pH8.0)で懸濁した。プロトプラスト懸濁液を200μlずつ15ml容の遠心チューブに移し、それぞれにSolution 2(40%(w/v) PEG♯4000、 50mM CaCl2、50mM Tris-HCl、pH8.0)40μlと前述の形質転換用DNA溶液各5μl(DNA量として5μg)を加えよく混合し、氷中で30分間放置した。1 mlのSol.2を加え混合し、室温で20分間放置した。5mlのSol.1で2回洗浄し、Sol.2をなるべく取り除いた。50℃に温めておいた終濃度0.02μg/mlのビオチン、5mMのウリジン、10mMのウラシル、0.5μg/mlのピリドキシンを添加したCzapek-Dox(CD)軟寒天培地にプロトプラスト縣濁液を加え混合し、終濃度0.02μg/mlのビオチン、5mMのウリジン、10mMのウラシル、0.5μg/mlのピリドキシンを添加したCD寒天培地に重層した。その後、30℃で分生子を形成するまで培養した。形質転換体からのmpkBΔ株の選択は以下のプライマー(5'-ATGCACTGTAAACTTTCGATCCCGTCTTAG-3'(配列番号211)、5'-CTACAGGGTGTGACGCTATGGTCAG-3'(配列番号212))を用いて形質転換体のゲノムDNAに対してPCRを行い、約4,700bpの増幅断片のみがみられたものをmpkBΔ候補株とし、最終的に定量RT-PCRによりmpkB遺伝子が発現していないことを確認した(図10)。
【0127】
(3)mpkBΔ株におけるマイクロアレイ解析
アスペルギルス・ニドランスABPU1 ΔligD::ptrA株及びmpkB破壊株の分生子を2x106個分生子/mlになるように、終濃度0.02μg/mlのビオチン、5mMのウリジン、10mMのウラシル、0.5μg/mlのピリドキシンを添加した200mlのCD液体培地を入れた500ml容の三角フラスコに接種し、37℃、24時間振盪培養した。菌体をMIRACLOTH(CALBIOCHEM社製)で集菌し、この回収した菌を乳鉢中で液体窒素を注ぎながらパウダー状になるまで粉砕した。10 mlのSepasol-RNA I Super(ナカライテスク社製)を入れた50 ml容チューブにパウダー状の菌体を移し懸濁後、室温にて5分間放置した。9,500×g、4℃、10分間遠心し、水層を15ml容チューブに移し、2 mlのクロロホルムを加え激しく撹拌後、室温にて5分間放置した。11,000×g、4℃、15分間遠心し、水層を別の15ml容チューブに移し、等量の水飽和酸性フェノール:クロロホルム(5:1)を加え激しく撹拌し、室温にて5分間放置した。10,000×g、4℃、10分間遠心し、水層を別の15ml容チューブに移し、半量のイソプロパノール、半量のHigh Salt Precipitation Solution(0.8M Sodium Citrate、1.2M NaCl、DEPC処理済み)を加え、撹拌後、室温にて10分間放置した。10,000×g、4℃、10分間遠心し、上清を捨て、70%エタノールでリンスした。風乾後、適量のDEPC(diethylpyrocarbonate)水に溶解し、total RNA溶液とした。次にoligotex-dt30 〈Super〉mRNA Purification kit (From Total RNA)(タカラバイオ社製)を用い、添付のマニュアルに従いtotal RNAからmRNAを精製した。
【0128】
マイクロアレイ解析をするにあたりプローブの作製は、Cy3、Cy5を用いたポストラベリング法でおこなった。精製したmRNAを1μg/9μlに調製し、CyScribe cDNA Post Labelling Kit(GE healthcare)を用い、添付のマニュアルに従ってmRNAからアミノアリル標識cDNAを合成した。合成したアミノアリル標識cDNAはCyScribe GFX Purification Kit(GE healthcare)を用いて精製し、続いてCyScribe cDNA Post Labelling Kit(GE healthcare)によりCyDye標識した。この時、ABPU1 ΔligD::ptrA株由来のcDNAをCy3で、mpkB破壊株由来のcDNAをCy5で標識した組と、その逆(Dyeスワップ)で標識した組とを準備した。標識したプローブはCyScribe GFX Purification Kit(GE healthcare)により精製し、遠心エバポレーターで乾燥した。表17に記すハイブリダイゼーション溶液を調製し、この溶液60μLを用いて上記組み合わせになるように、Cy3標識したcDNAを溶解させた後、その溶解液を用いてCy5で標識したcDNAを溶解させるといった手順で混合しハイブリダイゼーション溶液とした。
【0129】
【表17】
【0130】
アスペルギルス・ニドランスのマイクロアレイは、TIGERのガラススライドマイクロアレイ(version2)を使用した。プレハイブリダイゼーションバッファー(5×SSC、0.1% SDDS、1%BSA)を調製し、このバッファー中にガラススライドを浸し、少なくとも1時間、42℃で保温した。ガラススライドをMilliQ水で満たした洗浄用容器中に移し、2分間室温にて振盪洗浄した。MilliQ水での洗浄はMilliQ水2Lを使い切るまで繰り返した。次に別の容器にイソプロパノールを満たし、これにガラススライドを移して、2分間室温にて振盪洗浄した。その後800rpm、2分間遠心しイソプロパノールを完全に除去した。
【0131】
プレハイブリダイゼーション後のアレイスライドにSpaced Cover Glass XL (タカラバイオ社製)を被せ、アレイスライドとカバーガラスの間に前述の標識したハイブリダイゼーション溶液を充填させた。これをSlideBooster(Advaritix)に設置し、42℃で17時間保持した。その後SlideBoosterからアレイスライドを取り出し、Wash buffer 1(2×SSC、0.1% SDS)を満たした洗浄用容器中でカバーガラスを剥がした。アレイスライドをWash buffer 1中で15分間室温にて振盪洗浄した。次にWash Buffer 2(1×SSC、0.1% SDS)を満たした洗浄用容器に移し、5分間室温にて振盪洗浄した。最後にWash Buffer 3(0.2×SSC)を満たした洗浄用容器に移し、5分間室温にて振盪洗浄した後、800rpm、2分間遠心し水分を完全に除去した。
【0132】
アレイスライドの画像化にはGenePix4000B(Axon)を使用した。画像化したスキャンデータをGenePix4000Bに標準添付されているGenePixProマイクロアレイ解析ソフトを用いて数値化した。アレイデータはGenomic Profiler(三井情報(株))を用いて標準化し、Excelによりt検定をおこなった。ここで得られたデータをt検定のp値により絞り込むことによって、十分信頼性のおけるデータのみ後の解析に使用した。得られたアレイデータから、アスペルギルス・ニドランスABPU1 ΔligD::ptrA株とmpkB破壊株の間の発現量比の差が大きい遺伝子を抽出し、MpkB依存的に発現変動する遺伝子とした。
【0133】
〔実施例11〕
mpkBΔ株におけるsodM遺伝子の定量RT-PCR
(1)RNAの抽出
アスペルギルス・ニドランスABPU1 ΔligD::ptrA株及びmpkB破壊株の分生子を2x106個分生子/mlになるように、終濃度0.02μg/mlのビオチン、5mMのウリジン、10mMのウラシル、0.5μg/mlのピリドキシンを添加した100mlのCD液体培地を入れた300ml容の三角フラスコに接種し、37℃、24時間振盪培養した。菌体をMIRACLOTH(CALBIOCHEM社製)で集菌した。この回収した菌を乳鉢中で液体窒素を注ぎながらパウダー状になるまで粉砕した。10mlのSepasol-RNA I Super(ナカライテスク社製)を入れた50ml容チューブにパウダー状の菌体を移し懸濁後、室温にて5分間放置した。9,500×g、4℃、10分間遠心し、水層を15ml容チューブに移し、2mlのクロロホルムを加え激しく撹拌後、室温にて5分間放置した。11,000×g、4℃、15分間遠心し、水層を別の15ml容チューブに移し、等量の水飽和酸性フェノール:クロロホルム(5:1)を加え激しく撹拌し、室温にて5分間放置した。10,000×g、4℃、10分間遠心し、水層を別の15ml容チューブに移し、半量のイソプロパノール、半量のHigh Salt Precipitation Solution(0.8M Sodium Citrate、1.2M NaCl、DEPC処理済み)を加え、撹拌後、室温にて10分間放置した。10,000×g、4℃、10分間遠心し、上清を捨て、70%エタノールでリンスした。風乾後、適量のDEPC(diethylpyrocarbonate)水に溶解し、total RNA溶液とした。
【0134】
(2)cDNAの合成
cDNAの合成には、total RNAを用いた。260nmにおける吸光度からの計算値で100μgのtotal RNAをRNase freeの1.5mlチューブに移し、さらにDEPC処理水を加え87.5μlとした。これにRNase-free DNaseI set(QIAGEN)に付属のBuffer RDDを10μlとDNaseI solutionを10μl加え、37℃で10分間反応させて混荏するゲノムDNAの分解を行った。その後、RNeasy mini kit(QIAGEN)のRNA cleanupプロトコールに従ってtotal RNAをクリーンアップした。クリーンアップしたtotal RNA 2μg量をRNase freeの1.5mlチューブに移し、DEPC処理水を加え10μlとした。これにHigh Capacity cDNA Reverse Transcription kit(Applied Biosystems)に付属の反応液を表18に示す容量加え、20μlのcDNA合成反応液を調製した。
【0135】
【表18】
【0136】
調製したcDNA合成反応液を25℃で2分間保温し、プライマーをアニーリングさせた。その後、37℃で120分間保温し、逆転写反応させた。次に85℃で5秒間保温することにより、Multiscribe Reverse Transcriptaseを失活させた。反応終了後のcDNA溶液を1/10希釈し、次の定量RT-PCR実験に供した。
【0137】
(3)定量RT-PCR
合成したcDNAの蛍光色素によるラベリング反応には、DyNAmo SYBR Green qPCR Kit(FinnzymeS社製)を用いた。スキャナー解析及びデータ解析には、MiniOpticonリアルタイムPCR解析システム(バイオ・ラッドラボラトリーズ(株))を用いた。ラベリング反応操作は、DyNAmo SYBR Green qPCR Kit添付マニュアルにしたがった。通常の生育条件でのmpkBΔ株とコントロール株におけるsodM遺伝子の転写量の変化を調べた。コントロールをHistone H2Bとし、それぞれ以下の特異的プライマーを用いた。
Histone H2B FW:5'-CACCCGGACACTGGTATCTC-3'(配列番号213)
Histone H2B RW:5'-GAATACTTCGTAACGGCCTTGG-3'(配列番号214)
sodM FW:5'-CTCTGTAGAGGCGTTCATCAAG-3'(配列番号215)
sodM RW:5'-ACTGCAAGTAATACGCATGCTC-3'(配列番号216)
【0138】
1.5mlチューブに、5μlのcDNA(cDNA合成時に用いたtotalRNA量で100ng相当)、300nMの各標的遺伝子特異プライマー、10μlの2xMasterMix、滅菌水を加え25μlとした。専用のPCRチューブ8-Strip Low Profile Tubesに分注し、8-Strip Ultra Clear Caps(共にMJResearch社)にてふたをし、MiniOpticonリアルタイムPCR解析システムにセットした。反応条件は95℃、10分間のヒートショック後、熱変性95℃、10秒、アニーリング60℃、30秒、伸長反応72℃、30秒、プレートリードの反応を40サイクル行い、60〜95℃のグラジェント(0.2℃おきに1秒間hold)でMelting Curveの作成を行った。設定はHistone H2Bをスタンダード、sodMをサンプルとして設定した。PCRは各cDNA、各標的遺伝子につき三連で行った。PCRを行うにあたり、各標的遺伝子特異プライマー対と2×Master Mixのみの反応系を組み、95℃、10分間のヒートショック後、熱変性95℃、10秒、60〜95℃のグラジェントで50秒、プレートリードの反応を40サイクル行い、50〜95℃のグラジェントで(0.5℃おきに2秒間hold)でMelting Curveの作成を行い、プライマー同士の非特異的な増幅が起きないことを確認した。PCR終了後、MiniOpticonリアルタイムPCR解析システム付属の専用解析ソフトを用いて解析を行った。Quantitation画面上で各遺伝子のPCR産物が対数的に増幅し始めるサイクル数(CT)を決定した。この値をもとに各cDNAの標的遺伝子の相対発現量を以下の計算方法から求めた。
a)△CT値の算出(各cDNAの標的遺伝子とHistone H2Bとの差)
△CT=(y−x)
xはCT(Histone H2B)であり、yはCT(Target gene)である。
b)△CT値の算出(あるcDNAの△CTと基準とするcDNAの△CTとの差)
△CT=△CT(C)−△CT(S)
CはComparative cDNAであり、SはStandard cDNAである。
C)相対発現量の算出
2-(△CT)
また、Melting Curveの波形から正しく増幅されていることを確認し、さらにPCR産物をアガロースゲル電気泳動に供し、目的の大きさに増幅されていることを確認した。
【0139】
RT-PCRの結果を図10に、Histone H2Bとの発現量比としてグラフ化したものを示した。mpkBΔ株におけるsodM遺伝子の発現量はコントロール株と比べて優位に増加した。すなわち、sodM遺伝子の転写量の変化はMpkBに依存していると考えられた。よって、sodM遺伝子の5'-UTR(5'上流領域のプロモーター)はレポーター遺伝子と機能的に結合させることによって、本発明のスクリーニング方法において、被検物質のMpkB経路の阻害作用を検出することができる。
【0140】
〔実施例12〕
アスペルギルス・オリゼ(Aspergillus oryzae)のMpkA依存的遺伝子の決定
(1)RNA抽出
アスペルギルス・オリゼのmpkA遺伝子破壊株(NS4 ΔligD::ptrA ΔmpkA::sC)及びコントロール株(NS4 ΔligD::ptrA sC+)は酒類総合研究所の水谷治博士より供与いただいた(Mizutani et al. Fungal Genet. Biol., 2008 (45) 878-889)。mpkA遺伝子破壊株及びコントロール株の分生子を2×106個分生子/mlになるように、窒素源をグルタミン酸ナトリウムに置き換えた200 mlのCD液体培地を入れた500 ml容の三角フラスコに接種し、30℃、24時間振盪培養した。
【0141】
菌体をMIRACLOTH(CALBIOCHEM社製)で集菌し、この回収した菌を乳鉢中で液体窒素を注ぎながらパウダー状になるまで粉砕した。10mlのSepasol-RNA I Super(ナカライテスク)を入れた50ml容チューブにパウダー状の菌体を移し懸濁後、室温にて5分間放置した。9,500×g、4℃、10分間遠心し、水層を15ml容チューブに移し、2mlのクロロホルムを加え激しく撹拌後、室温にて5分間放置した。11,000×g、4℃、15分間遠心し、水層を別の15ml容チューブに移し、等量の水飽和酸性フェノール:クロロホルム(5:1)を加え激しく撹拌し、室温にて5分間放置した。10,000×g、4℃、10分間遠心し、水層を別の15ml容チューブに移し、半量のイソプロパノール、半量のHigh Salt Precipitation Solution(0.8M Sodium Citrate、1.2M NaCl、DEPC処理済み)を加え、撹拌後、室温にて10分間放置した。10,000×g、4℃、10分間遠心し、上清を捨て、70%エタノールでリンスした。風乾後、適量のDEPC(diethylpyrocarbonate)水に溶解し、total RNA溶液とした。次にoligotex-dt30 〈Super〉mRNA Purification kit (From Total RNA)(タカラバイオ社製)を用い、添付のマニュアルに従いtotal RNAからmRNAを精製した。
【0142】
マイクロアレイ解析をするにあたりプローブの作製は、Cy3、Cy5を用いたポストラベリング法でおこなった。精製したmRNAを1μg/9μlに調製し、CyScribe cDNA Post Labelling Kit(GE healthcare社製)を用い、添付のマニュアルに従ってmRNAからアミノアリル標識cDNAを合成した。合成したアミノアリル標識cDNAはCyScribe GFX Purification Kit(GE healthcare社製)を用いて精製し、続いてCyScribe cDNA Post Labelling Kit(GE healthcare社製)によりCyDye標識した。この時、コントロール株由来のcDNAをCy3で、mpkA破壊株由来のcDNAをCy5で標識した組と、その逆(Dyeスワップ)で標識した組とを準備した。標識したプローブはCyScribe GFX Purification Kit(GE healthcare社製)により精製し、遠心エバポレーターで乾燥した。実施例10の表17に示したハイブリダイゼーション溶液を調製し、この溶液60μLを用いて上記組み合わせになるように、Cy3標識したcDNAを溶解させた後、その溶解液を用いてCy5で標識したcDNAを溶解させるといった手順で混合しハイブリダイゼーション溶液とした。
【0143】
アスペルギルス・オリゼのマイクロアレイは、ファームラボ社製の麹菌12Kガラススライドマイクロアレイを使用した。アレイスライドにギャップカバーガラス(マツナミ社製)を被せ、アレイスライドとカバーガラスの間に前述の標識したハイブリダイゼーション溶液を充填させた。アレイスライドチャンバー(タカラバイオ社製)に設置し、40℃に設定したウォーターバス中で17時間保持した。その後アレイスライドチャンバーからアレイスライドを取り出し、Wash buffer 1(2 ×SSC、0.03% SDS)を満たしたプラスチック製の洗浄用容器中でカバーガラスを剥がした。アレイスライドをWash buffer 1中で15分間室温にて振盪洗浄した。次にWash Buffer 2(0.2× SSC)を満たした洗浄用容器に移し、5分間室温にて振盪洗浄した。最後にWash Buffer 3(0.05 × SSC)を満たした洗浄用容器に移し、5分間室温にて振盪洗浄した後、800rpm、2分間遠心し水分を完全に除去した。
【0144】
アレイスライドの画像化にはGenePix4000B(Axon)を使用した。画像化したスキャンデータをGenePix4000Bに標準添付されているGenePixProマイクロアレイ解析ソフトを用いて数値化した。アレイデータはGenomic Profiler(三井情報(株))を用いて標準化し、Excelによりt検定をおこなった。ここで得られたデータをt検定のp値により絞り込むことによって、十分信頼性のおけるデータのみ後の解析に使用した。得られたアレイデータから、アスペルギルス・オリゼmpkA破壊株とコントロール株の間の発現量比の差が大きい遺伝子を抽出し、MpkA依存的に発現変動する遺伝子とした。
【0145】
その結果、アスペルギルス・オリゼのMpkA依存的に発現変動する遺伝子のうち、経路遮断により活性上昇する遺伝子(AO090038000214、AO090003001496、 AO090009000634、AO090102000339)、及び経路遮断により活性が抑制される遺伝子(AO090003001367、AO090012000623、AO090012000169、AO090012000625、AO090012000622、AO090003001336、AO09001000644)が特定された。これら遺伝子のプロモータは、レポーター遺伝子と機能的に結合させることによって、MAPキナーゼ経路の一つであるMpkA経路に対する被検物質の活性化作用又は阻害作用を検出することができる。
【0146】
〔実施例13〕
本実施例では、Fujioka et al.(2007) Eukaryotic Cell 6: 1497-1510 の論文中に記載されている、cell wall integrity 経路を構成するMAP キナーゼMpkAの破壊株と野生株における転写解析のデータをもとにagsA遺伝子及びagsB遺伝子のプロモータ領域を使用したレポーター株を作製した。
【0147】
(1)レポーター遺伝子ベクターの構築
Broad Institute のアスペルギルス属ゲノムデータベース(http://www.broad.mit.edu/annotation/genome/aspergillus_group/MultiHome.html)からアスペルギルス・ニドランス(Aspergillus nidulans)のagsA遺伝子及びagsB遺伝子の5'上流領域1,000 bpを表19に示すプライマーセットを用いてPCRで増幅した。
【0148】
【表19】
【0149】
PCR産物をpGEM-T Easy Vector(Promega社製)に導入し、pGEM-PagsA及びpGEM-PagsBを得た。次いでpGEM-PagsA及びpGEM-PagsBをNotIで消化し、PagsA及びPagsB(PagsA及びPagsBの5'上流領域1,000bP)を切り出し、pNA(N)EGFPのNotIサイトへ導入してpNA(N)EGFP/PagsA及びpNA(N)EGFP/PagsBを作製した。なおpNA(N)EGFPは東北大学大学院農学研究科・古川健太郎博士より供与して頂いたもので、EGFPに核移行型シグナルLacIが付加してある(Furukawa et al. Biosci. Biotechnol. Biochem., 2007 (71) 1724-1730)。
【0150】
上記プラスミドにおいて、pNA(N)EGFP/PagsAについてはオーレオバシジン耐性遺伝子aurA内に存在するSrfIサイトで、pNA(N)EGFP/PagsBについてはaurA内に存在するBssHIIサイトで直鎖状に切断した断片を用いて、A.nidulans A89+argB(正式にはRB89)株及びmpkAΔ株(A89 ΔmpkA::argB)に対して形質転換を行った。オーレオバシジン耐性株を選抜し、PCRにより遺伝子導入を確認した。
【0151】
(2)EGFP-LacI蛍光観察
96ウェルのタイタープレートにCDビオチン液体培地200μL、RB89株からの形質転換体から調製した胞子懸濁液20(200コ)の混合液を加え、30℃で24時間培養した。タイタープレートから菌体をこすり取りEGFP-LacIの蛍光を観察した。PagsB-EGFP形質転換体においては、核に局在したEGFP-LacI蛍光が観察されたが、PagsA-EGFP形質転換体では蛍光は観察されなかった。一方、mpkAΔ株を宿主とした場合には、PagsA-EGFP形質転換体では蛍光が観察され、PagsB-EGFP形質転換体では蛍光は観察されなかった(図11)。これはagsA、agsBの発現解析パターンと完全に一致するものである(Fujioka et al. Eukaryotic Cell, 2007 (6) 1497-1510)。以上の結果から、PagsA-EGFP及びPagsB-EGFPレポ一夕ーはMpkA経路のシグナル切断作動性薬剤のスクリーニングに利用できると言える。
【0152】
〔実施例14〕
cat1遺伝子のHOG経路レポーター遺伝子としての可能性検証
(1)ゲノムデータベースからのいもち病菌CAT-1遺伝子ホモログの探索
本実施例では、アカパンカビ(Neurospora crassa)においてCAT-1(catalase 1)遺伝子がHOG経路依存的に転写制御を受けるという報告に基づき、いもち病菌のゲノムデータベース (http://www.broad.mit.edu/annotation/genome/magnaporthe_grisea/MultiHome.html) からCAT-1遺伝子のオルソログを検索した。N. crassaのCAT-1遺伝子(NCU08791.3)をクエリー配列としてblastp検索を行ったところ、MGG_10061.6(Genbankアクセッション番号XM_365841)が高いホモロジーでヒットしたことからこの遺伝子をいもち病菌におけるcat1遺伝子とした。
【0153】
(2)total RNAの調製及びcDNAの合成
いもち病菌におけるcat1遺伝子がHOG経路構成的活性化剤フルジオキソニル又はイプロジオンを処理したときに、特異的に発現変化があり検出マーカーとして利用可能か否かを検証した。
【0154】
ブレンダーで細かく裁断したいもち病菌の菌体を50ml YG液体培地(0.4% yeast extract、2% glucose/200ml三角フラスコ)で27℃、120rpm、22h培養後、20mg/mlのフルジオキソニル(in DMSO)を50μl添加した(終濃度20μg/ml)。コントロールはDMSOを50μl添加した。薬剤添加後15分間培養を続け、ミラクロスで菌体を回収した。液体窒素を用いて菌体を破砕し5mlのTRIzol (Invitrogen社製)に懸濁後5分間静置した。1mlのクロロホルムを加え激しく攪拌後、室温で3分間静置した。5,000 rpm、15min、4℃で遠心し、RNAを含む約3mlの水層を別のチューブに移した。このRNA 画分のうち1.2mlをPureLink Micro-to-Midi Total RNA Purification Kit (Invitrogen社製)を用いて精製した。このtotal RNAをPrimeScript RT reagent Kit (Perfect Real Time)(タカラバイオ社製)を用いて逆転写し、一本鎖cDNAを作製した。逆転写のprimerにはOligo dT PrimerとRandom 6 mersの両方を用いた。
【0155】
(3)リアルタイムPCRによるcat1遺伝子の転写応答解析
内部標準の遺伝子にはβ-tubulin遺伝子(MGG_00604.6)を用いた。解析に用いたプライマーを以下に示す。
いもち病菌におけるcat1遺伝子用
Mgcat1 FW1: 5'-TGGATCAGTTCTTCCACAGCA-3'(配列番号221)
Mgcat1 RV1: 5'-AACTCCACCCGCTTTCTCC-3'(配列番号222)
β-tubulin遺伝子用
Mg_b-tubulin FW1: 5'-GAGTCCAACATGAACGATCT-3'(配列番号223)
Mg_b-tubulin RV1: 5'-GTACTCCTCTTCCTCCTCGT-3'(配列番号224)
【0156】
リアルタイムPCRの反応系の調製にはSYBR Premix Ex Taq II (Perfect Real Time) (タカラバイオ社製)を用い、thermal cyclerには Thermal Cycler Dice Real Time System (タカラバイオ社製)を使用した。反応条件は、初期変性95℃、30秒、その後、変性95℃、5秒、アニーリング/伸長:60℃、30秒(40サイクル)で行った。
【0157】
RT-PCRの結果を図12に示した。cat1遺伝子の転写量はフルジオキソニル及びイプロジオン処理により有意に増加していた。よって、いもち病菌におけるcat1遺伝子の5'-UTR(5'上流領域のプロモーター)はレポーター遺伝子と機能的に結合させることにより、HOG経路活性化剤のスクリーニング系として利用できる可能性が確認された。
【産業上の利用可能性】
【0158】
本発明は、感染性微生物に使用される抗菌剤を開発する分野に利用することができる。
【技術分野】
【0001】
本発明は、作用物質による刺激により特異的に発現する遺伝子をレポーター遺伝子として使用するスクリーニング方法及び当該遺伝子の発現を制御するプロモータに関する。
【背景技術】
【0002】
微生物は、高浸透圧、高温、栄養飢餓、酸化、創傷といった様々な環境ストレスに曝される。これら環境ストレスに微生物が曝されると、特定の遺伝子発現といった生化学的、生理学的及び分子的な反応が誘導され、環境ストレスに対する応答性及び順応性を示すことが知られている。このような反応は、微生物細胞内における刺激の認識及びシグナル伝達の結果として進行する。抗菌剤への暴露も環境ストレスの1つであり、特定の遺伝子発現が誘導されることが示唆されるが、抗菌剤の抗菌作用と、当該抗菌作用の付加時に特異的に発現誘導される遺伝子との関係は多くの知見がないのが現状である。
【0003】
例えば、呼吸は生物にとって生存に必須であり、呼吸鎖阻害剤は抗菌剤として高い効果を示す。したがって、公知の呼吸鎖阻害剤と同様の抗菌作用を示す物質について、更なる開発が望まれている。
【0004】
また、エルゴステロールは真菌に特有の細胞膜の成分で、生存に必須であり、エルゴステロール生合成阻害剤は抗菌剤として高い効果を示す。したがって、公知のエルゴステロール生合成阻害剤と同様の抗菌作用を示す物質について、更なる開発が望まれている。
【0005】
さらに、グルカン、キチンなどから成る細胞壁は真菌に特有の成分で、生存に必須であり、細胞壁生合成阻害剤は抗菌剤として高い効果を示す。したがって、公知の細胞壁生合成阻害剤と同様の抗菌作用を示す物質について、更なる開発が望まれている。
【0006】
さらにまた、cell wall integrity経路は、BckA(MAPKKK)→MkkA(MAPKK)→MpkA(MAPK)からなるMAP キナーゼシグナル伝達経路を中心とした細胞壁構築制御経路であり、真菌類に広く保存されている生体システムであり、細胞壁生合成阻害剤への暴露などにより細胞壁に損傷を受けると本経路が活性化し、細胞壁生合成関連遺伝子の転写を制御して細胞壁の再構築が行われることが知られている。cell wall integrity経路の遮断または構成的な活性化は細胞に重篤な障害を引き起こすことから、cell wall integrity経路阻害薬は抗菌剤として高い効果を示すと考えられる。したがって、cell wall integrity経路阻害薬の開発が望まれている。
【0007】
さらにまた、MpkB経路は、出芽酵母のSte11(MAPKKK)→Ste7(MAPKK)→Fus3/Kss1(MAPK)からなるMAP キナーゼシグナル伝達経路の相同経路で、真菌類に広く保存されている生体システムであり、多くの植物病原菌において、本経路が病原性に必須であることが報告されていることから、MpkB経路阻害薬は抗菌剤として高い効果を示すと考えられる。したがって、MpkB経路阻害薬の開発が望まれている。
【0008】
さらにまた、HOG経路は、出芽酵母のSsk2/22(MAPKKK)→Pbs2(MAPKK)→Hog1(MAPK)からなるMAP キナーゼシグナル伝達経路の相同経路で、真菌類に広く保存されている生体システムであり、多くの植物病原菌において、本経路が病原性に必須であることが報告されている。HOG経路の上流には、ヒスチジンキナーゼ→リン酸基仲介因子→レスポンスレギュレーターからなる二成分性情報伝達系が存在し、このヒスチジンキナーゼに作用してHOG経路を構成的に活性化することで抗菌活性を示す薬剤が農薬の有効成分として実用化もされており、HOG経路作動薬は抗菌剤として高い効果を示す。したがって、公知のHOG経路作動薬と同様の抗菌作用を示す物質について、更なる開発が望まれていることから、本経路を標的とする薬剤の開発が望まれている。
【発明の概要】
【発明が解決しようとする課題】
【0009】
そこで、本発明は、上述したような実情に鑑み、種々の抗菌作用を付加した際に特異的に発現する遺伝子を特定し、当該遺伝子をレポーター遺伝子として用いることで、公知の抗菌剤の候補物質或いは公知の抗菌剤と同等の抗菌作用を有する物質をスクリーニングする方法を提供することを目的とする。
【0010】
また、本発明は、種々の抗菌作用を付加した際に特異的に発現する遺伝子及び当該遺伝子の発現を制御するプロモータ領域を特定し、種々の抗菌作用を付加したときに発現を誘導するといった特徴を有するプロモータを提供することを目的とする。
【課題を解決するための手段】
【0011】
上述した目的を達成した本発明は以下を包含する。
本発明に係るスクリーニング方法は、被検物質を作用させた細胞における、表1に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる少なくとも1種の遺伝子の発現を測定する工程と、上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を呼吸鎖阻害剤の候補物質として同定する工程とを含んでいる。
【0012】
【表1】
【0013】
また、本発明に係るスクリーニング方法は、被検物質を作用させた細胞における、表2に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる少なくとも1種の遺伝子の発現を測定する工程と、上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質をエルゴステロール生合成阻害剤の候補物質として同定する工程とを含んでいる。
【0014】
【表2】
【0015】
さらに、本発明に係るスクリーニング方法は、被検物質を作用させた細胞における、表3に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる少なくとも1種の遺伝子の発現を測定する工程と、上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を細胞壁生合成阻害剤の候補物質として同定する工程とを含んでいる。
【0016】
【表3】
【0017】
さらにまた、本発明に係るスクリーニング方法は、被検物質を作用させた細胞における、スーパーオキシドデスムターゼ遺伝子(Genbankアクセッション番号:XM_653297、配列番号71)の発現を測定する工程と、上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質をMpkB経路阻害剤の候補物質として同定する工程とを含んでいる。
【0018】
さらにまた、本発明に係るスクリーニング方法は、被検物質を作用させた細胞における、表4に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる少なくとも1種の遺伝子の発現を測定する工程と、上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を細胞壁構築(cell wall integrity)経路阻害剤の候補物質として同定する工程とを含んでいる。
【0019】
【表4】
【0020】
さらにまた、被検物質を作用させた細胞における、agsA遺伝子(Genbankアクセッション番号:XM_658397、配列番号83)及び/又はagsB遺伝子(Genbankアクセッション番号:XM_655819、配列番号84)の発現を測定する工程と、上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を細胞壁構築(cell wall integrity)経路阻害剤の候補物質として同定する工程とを含んでいる。
【0021】
さらにまた、本発明に係るスクリーニング方法は、被検物質を作用させた細胞における、cat1遺伝子(Genbankアクセッション番号:XM_365841、配列番号85)の発現を測定する工程と、上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を浸透圧応答(HOG: high osmolarity glycerol)経路活性化剤の候補物質として同定する工程とを含んでいる。
【発明の効果】
【0022】
以上のように、本発明に係るスクリーニング方法によれば、呼吸鎖阻害剤の候補物質、エルゴステロール生合成阻害剤の候補物質、細胞壁生合成阻害剤の候補物質、MpkB経路阻害剤の候補物質、細胞壁構築(cell wall integrity)経路阻害剤の候補物質又は浸透圧応答(HOG: high osmolarity glycerol)経路活性化剤の候補物質を同定することができる。すなわち、本発明によれば、上記の各種抗菌作用に対する簡便な評価系を確立することができ、多数の化合物群からなる薬剤ライブラリーから候補物質を、高速かつ高効率で同定することができる。
【図面の簡単な説明】
【0023】
【図1】シアン耐性呼吸酵素遺伝子(AOX)(上段)及びNADH-デヒドロゲナーゼ遺伝子(下段)についてRT-PCRにより測定した発現量をactin当たりの発現量として示す特性図である。
【図2】糸状菌形質転換ベクターpCH11の概略構成図である。
【図3】pCH11-EGFP-LacIの概略構成図である。
【図4】pCH11-PrAOX-EGFP-LacIプラスミドを導入したマグナポルテ・グリセアに対して、呼吸鎖阻害剤及び比較薬剤を処理した時のEGFPの蛍光を観察した結果を示す特性図である。
【図5】Cytochrome P450ステロール14αジメチラーゼ(MGG_04628、上段)及びステロール24Cメチルトランスフェラーゼ(MGG_10860、下段)についてRT-PCRにより測定した発現量をactin当たりの発現量として示す特性図である。
【図6】pCH11- Cyp51-EGFP-LacIプラスミドを導入したマグナポルテ・グリセアに対して、エルゴステロール生合成阻害剤及び比較薬剤を処理した時のEGFPの蛍光を観察した結果を示す特性図である。
【図7】MGG_02004(上段の左)、MGG_00692(上段の右)、MGG_04943(中段の左)、MGG_09639(中段の右)、MGG_05133(下段の左)及びMGG_02773(下段の右)についてRT-PCRにより測定した発現量をactin当たりの発現量として示す特性図である。
【図8】pNA(N)EGFPの概略構成図である。
【図9】pMSTU1-EGFP及びpAGS1-EGFPを導入したマグナポルテ・グリセアに対して、細胞壁生合成阻害剤を処理した時のEGFPの蛍光を観察した結果を示す特性図である。
【図10】mpkBΔ株におけるsodM遺伝子の発現量をHistone H2B当たりの発現量として示す特性図である。
【図11】PagsB-EGFP形質転換体及びPagsA-EGFP形質転換体について、蛍光を観察した結果を示す特性図である。
【図12】いもち病菌におけるcat1遺伝子についてRT-PCRにより測定した発現量をβ-tubulin当たりの発現量として示す特性図である。
【発明を実施するための形態】
【0024】
以下、本発明をより詳細に説明する。
本発明に係るスクリーニング方法は、種々の被検物質の中から、呼吸鎖阻害剤の候補物質、エルゴステロール生合成阻害剤の候補物質、細胞壁生合成阻害剤の候補物質、MpkB経路阻害剤の候補物質、細胞壁構築(cell wall integrity)経路阻害剤の候補物質又は浸透圧応答(HOG: high osmolarity glycerol)経路活性化剤の候補物質を同定する方法である。ここで、被験物質としては、特に限定されず如何なる物質であってもよい。被験物質としては、単独の物質であってもよいし、複数の構成成分からなる混合物であってもよい。被験物質としては、例えば植物からの抽出物のように未同定の物質を含むような構成であってもよいし、既知の組成物を所定の組成比で含むような構成であってもよい。また、被験物質としては、タンパク質、核酸、脂質、多糖類、有機化合物及び無機化合物のいずれでもよい。
【0025】
また、本明細書において、呼吸鎖阻害剤とは、微生物、より好ましくは植物または動物への感染性微生物における呼吸鎖を阻害する抗菌作用を有する物質を意味する。また、エルゴステロール生合成阻害剤とは、真菌、より好ましくは植物または動物への感染性真菌におけるエルゴステロール生合成を阻害する抗菌作用を有する物質を意味する。細胞壁生合成阻害剤とは、真菌、より好ましくは植物または動物への感染性真菌における細胞壁生合成を阻害する抗菌作用を有する物質を意味する。MpkB経路阻害剤とは、真菌、より好ましくは植物または動物への感染性真菌におけるMpkBが関与するMAPキナーゼシグナル伝達経路(MpkB経路)を阻害する抗菌作用を有する物質を意味する。細胞壁構築経路阻害剤とは、細胞壁の損傷時に活性化するcell wall integrity経路を阻害する抗菌作用を有する物質を意味する。HOG経路活性化剤とは、HOG経路を活性化することで抗菌作用を有する物資を意味する。
【0026】
本発明に係るスクリーニング方法により同定された候補物質は、その抗菌作用により種々の微生物、好ましくは感染性微生物に対する抗菌剤として使用することができる。感染性微生物としては例えば、いもち病菌(Magnaporthe grisea)を挙げることができる。すなわち、本発明に係るスクリーニング方法によれば、いもち病菌(Magnaporthe grisea)等の感染性微生物に対する抗菌剤の候補物質を同定することができる。
【0027】
本発明に係るスクリーニング方法は、呼吸鎖阻害剤の指標となる遺伝子、エルゴステロール生合成阻害剤の指標となる遺伝子、細胞壁生合成阻害剤の指標となる遺伝子、MpkB経路阻害剤の指標となる遺伝子、細胞壁構築(cell wall integrity)経路阻害剤の指標となる遺伝子、及びHOG経路活性化剤の指標となる遺伝子を使用した方法(スクリーニング方法1)並びにこれら遺伝子の発現を制御するプロモータ領域及び公知のレポーター遺伝子を使用した方法(スクリーニング方法2)に大別される。
【0028】
まず、これら抗菌剤の指標となる遺伝子について説明する。
呼吸鎖阻害剤の指標となる遺伝子
呼吸鎖阻害剤の指標となる遺伝子は、従来公知の呼吸鎖阻害剤をいもち病菌(Magnaporthe grisea)に接触させた時に特異的に発現が亢進する遺伝子として見いだされた遺伝子である。ここで従来公知の呼吸鎖阻害剤とは、特に限定されないが、ジフルメトリム及びテブフェンピラド等のComplex1阻害剤、メプロニル等のComplex2阻害剤、クレソキシムメチル及びピリベンカルブ等のComplex3阻害剤、オリゴマイシン等のComplex5(F1Fo ATPase)阻害剤、フルアジナム等の電子伝達に作用してATP合成を阻害する脱共役作用剤を挙げることができる。
【0029】
呼吸鎖阻害剤の指標となる遺伝子は、下記表5に列挙したGenbankアクセッション番号によって特定される遺伝子である。下記表5における「CDS塩基配列」の欄には、Genbankアクセッション番号で特定される遺伝子のCDS領域の塩基配列を示す配列番号を記載している。また、下記表5における「BROAD ID Ver6」の欄には、Broad Instituteのマグナポルテ・グリセア(Magnaporthe grisea)ゲノムデータベースの登録番号を記載している。
【0030】
【表5】
【0031】
エルゴステロール生合成阻害剤の指標となる遺伝子
エルゴステロール生合成阻害剤の指標となる遺伝子は、従来公知のエルゴステロール生合成阻害剤をいもち病菌(Magnaporthe grisea)に接触させた時に特異的に発現が亢進する遺伝子として見いだされた遺伝子である。ここで従来公知のエルゴステロール生合成阻害剤とは、特に限定されないが、オキスポコナゾール・フマル酸塩及びフェンヘキサミドを挙げることができる。
【0032】
エルゴステロール生合成阻害剤の指標となる遺伝子は、下記表6に列挙したGenbankアクセッション番号によって特定される遺伝子である。下記表6における「CDS塩基配列」の欄には、Genbankアクセッション番号で特定される遺伝子のCDS領域の塩基配列を示す配列番号を記載している。また、下記表6における「BROAD ID Ver6」の欄には、Broad Instituteのマグナポルテ・グリセア(Magnaporthe grisea)ゲノムデータベースの登録番号を記載している。
【0033】
【表6】
【0034】
細胞壁生合成阻害剤の指標となる遺伝子
細胞壁生合成阻害剤の指標となる遺伝子は、従来公知の細胞壁生合成阻害剤をいもち病菌(Magnaporthe grisea)に接触させた時に特異的に発現が亢進する遺伝子として見いだされた遺伝子である。ここで従来公知の細胞壁生合成阻害剤とは、特に限定されないが、ミカファンギン及びカルコフロール・ホワイトを挙げることができる。
【0035】
細胞壁生合成阻害剤の指標となる遺伝子は、下記表7に列挙したGenbankアクセッション番号によって特定される遺伝子である。下記表7における「CDS塩基配列」の欄には、Genbankアクセッション番号で特定される遺伝子のCDS領域の塩基配列を示す配列番号を記載している。また、下記表7における「BROAD ID Ver6」の欄には、Broad Instituteのマグナポルテ・グリセア(Magnaporthe grisea)ゲノムデータベースの登録番号を記載している。
【0036】
【表7】
【0037】
MpkB経路阻害剤の指標となる遺伝子
MpkB経路阻害剤の指標となる遺伝子は、アスペルギルス・ニドランス(Aspergillus nidulans)の野生株と、mpkB遺伝子破壊株とにおける遺伝子の発現パターンを測定し、mpkB遺伝子破壊株において特異的に発現が亢進する遺伝子として見いだされた遺伝子である。
MpkB経路阻害剤の指標となる遺伝子は、スーパーオキシドデスムターゼ遺伝子(Genbankアクセッション番号:XM_653297、配列番号71)である。
【0038】
細胞壁構築経路阻害剤の指標となる遺伝子
細胞壁構築経路阻害剤の指標となる遺伝子は、アスペルギルス・オリゼ(Aspergillus oryzae)の野生株と、cell wall integrity経路に関与するmpkA遺伝子破壊株とにおける遺伝子の発現パターンを測定し、mpkA遺伝子破壊株において特異的に発現が亢進する遺伝子又は特異的に発現が低減する遺伝子として見いだされた遺伝子である。なお、mpkA遺伝子を破壊した株では、cell wall integrity経路が阻害されることとなる。
【0039】
細胞壁構築経路阻害剤の指標となる遺伝子は、下記表8に列挙したGenbankアクセッション番号によって特定される遺伝子である。下記表8における「CDS塩基配列」の欄には、Genbankアクセッション番号で特定される遺伝子のCDS領域の塩基配列を示す配列番号を記載している。また、下記表8における「DOGAN ID」の欄には、独立行政法人 製品評価技術基盤機構のDOGANデータベースの登録番号を記載している。
【0040】
【表8】
【0041】
なお、表8のうち、XM_001825115、XM_001820488、XM_001817008及びXM_001822481の遺伝子は、mpkA遺伝子破壊株において特異的に発現が亢進する遺伝子である。また、表8のうち、XM_001820375、XM_001727556、XM_001727176、XM_001727558、XM_001727555、XM_001820346及びXM_001827554の遺伝子は、mpkA遺伝子破壊株において特異的に発現が低減する遺伝子である。
【0042】
また、細胞壁構築経路阻害剤の指標となる遺伝子は、Fujioka et al.(2007) Eukaryotic Cell 6: 1497-1510 の論文中に記載されている、アスペルギルス・オリゼ(Aspergillus oryzae)のMpkA破壊株と野生株における転写解析のデータに基づいて選択したagsA遺伝子(Genbankアクセッション番号:XM_658397、配列番号83)及びagsB遺伝子(Genbankアクセッション番号:XM_655819、配列番号84)である。
【0043】
HOG経路活性化剤の指標となる遺伝子
HOG経路活性化剤の指標となる遺伝子は、アカパンカビ (Neurospora crassa)に関する研究でHOG経路の活性化により発現亢進することが知られるカタラーゼ遺伝子に対する、いもち病菌における相同遺伝子として見いだされた遺伝子である。また、当該相同遺伝子は、フルジオキソニル及びイプロジオン等の公知のHOG経路活性化剤に接触させた時に特異的に発現が亢進することを確認している。
HOG経路活性化剤の指標となる遺伝子cat1遺伝子(Genbankアクセッション番号:XM_365841、配列番号85)である。
【0044】
以上のように、異なる抗菌作用を有する抗菌剤の指標となる遺伝子を、Genbankアクセッション番号及び具体的な配列番号によって特定して説明したが、抗菌剤の指標となる遺伝子はCDS領域の塩基配列が上記配列番号に記載された具体的な塩基配列からなるものに限定されるものではなく、当該遺伝子に対する相同遺伝子も含む意味である。相同遺伝子とは、一般的に、共通の祖先遺伝子から進化分岐した遺伝子を意味しており、2種類の種の相同遺伝子(オルソログ(ortholog))及び同一種内で重複分岐により生じた相同遺伝子(パラログ(paralog))を含む意味である。
【0045】
より具体的には、上記配列番号に示す塩基配列に対して70%以上、好ましくは80%以上、さらに好ましくは90%以上、より好ましくは95%以上、最も好ましくは98%以上の類似度(Similarity)を有する塩基配列を含むCDS領域を有する遺伝子も、抗菌剤の指標となる遺伝子に含まれる。ここで、類似度の値は、BLAST(Basic Local Alignment Search Tool)プログラムを実装したコンピュータプログラム及び遺伝子配列情報を格納したデータベースを用いてデフォルトの設定で求められる値を意味する。
【0046】
また、より具体的には、上記配列番号に示す塩基配列に対して相補的な塩基配列を有するポリヌクレオチドにストリンジェントな条件下でハイブリダイズするcDNA又はゲノム領域から特定される遺伝子も、抗菌剤の指標となる遺伝子に含まれる。ここで、ストリンジェントな条件とは、いわゆる特異的なハイブリッドが形成され、非特異的なハイブリッドが形成されない条件をいう。例えば、45℃、6×SSC(塩化ナトリウム/クエン酸ナトリウム)でのハイブリダイゼーション、その後の50〜65℃、0.2〜1×SSC、0.1%SDSでの洗浄が挙げられ、或いはそのような条件として、65〜70℃、1×SSCでのハイブリダイゼーション、その後の65〜70℃、0.3×SSCでの洗浄を挙げることができる。ハイブリダイゼーションは、J. Sambrook et al. Molecular Cloning, A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory(1989)に記載されている方法等、従来公知の方法で行うことができる。
【0047】
スクリーニング方法1
次に、上述したスクリーニング方法1について説明する。被検物質が上述した抗菌作用を有するか評価するには、先ず、供試された被検物質を例えば感染性微生物に接触させ、被検物質の存在下における上記遺伝子の発現量と、被検物質の非存在下における発現量とを比較する。そして、公知の抗菌剤(呼吸鎖阻害剤、エルゴステロール生合成阻害剤、細胞壁生合成阻害剤、HOG経路活性化剤)を作用させた時と同様に又はmpkB遺伝子破壊株若しくはmpkA遺伝子破壊株と同様に、当該所定の遺伝子の発現量が変動している場合、当該被検物質は、上述した抗菌作用を有するものとして同定することができる。
【0048】
上述した遺伝子の発現量は、従来公知の手法により測定することができる。遺伝子の発現量を測定する手法としては、何ら限定されないが、例えばリアルタイム PCR法を適用することができる。また、マイクロアレイを用いて上述した遺伝子の発現量を測定しても良い。
【0049】
例えば、表5に示したGenbankアクセッション番号によって特定される遺伝子のうち少なくとも1以上、好ましくは5以上、より好ましくは10以上の遺伝子について、被検物質の存在下における発現量が亢進していた場合には、当該被検物質を呼吸鎖阻害剤の候補物質として同定することができる。特に、呼吸鎖阻害剤の候補物質を同定する場合、表5に示した遺伝子のうちXM_001403469で特定される遺伝子の発現量を検討することが好ましい。XM_001403469で特定される遺伝子について被検物質の存在下における発現量が亢進していた場合には、当該被検物質を呼吸鎖阻害剤の候補物質としての蓋然性が非常に高いと判断できる。
【0050】
また、例えば、表6に示したGenbankアクセッション番号によって特定される遺伝子のうち少なくとも1以上、好ましくは3以上、より好ましくは5以上の遺伝子について、被検物質の存在下における発現量が亢進していた場合には、当該被検物質をエルゴステロール生合成阻害剤の候補物質として同定することができる。特に、エルゴステロール生合成阻害剤の候補物質を同定する場合、表6に示した遺伝子のうちXM_362183で特定される遺伝子の発現量を検討することが好ましい。XM_362183で特定される遺伝子について被検物質の存在下における発現量が亢進していた場合には、当該被検物質をエルゴステロール生合成阻害剤の候補物質としての蓋然性が非常に高いと判断できる。
【0051】
さらに、例えば、表7に示したGenbankアクセッション番号によって特定される遺伝子のうち少なくとも1以上、好ましくは5以上、より好ましくは10以上、更に好ましくは20以上の遺伝子について、被検物質の存在下における発現量が亢進していた場合には、当該被検物質を細胞壁生合成阻害剤の候補物質として同定することができる。特に、細胞壁生合成阻害剤の候補物質を同定する場合、表7に示した遺伝子のうちXM_364794で特定される遺伝子及び/又はXM_368552で特定される遺伝子の発現量を検討することが好ましい。XM_364794で特定される遺伝子又はXM_368552で特定される遺伝子について被検物質の存在下における発現量が亢進していた場合には、当該被検物質を細胞壁生合成阻害剤の候補物質としての蓋然性が非常に高いと判断できる。
【0052】
さらにまた、例えば、Genbankアクセッション番号XM_653297で特定されるスーパーオキシドデスムターゼ遺伝子について被検物質の存在下における発現量が亢進していた場合には、当該被検物質をMpkB経路阻害剤の候補物質として同定することができる。
【0053】
さらにまた、例えば、表8に示したGenbankアクセッション番号によって特定される遺伝子のうち少なくとも1以上、好ましくは3以上、より好ましくは5以上の遺伝子について、被検物質の存在下における発現量が変動していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質として同定することができる。より具体的に、XM_001825115で特定される遺伝子、XM_001820488で特定される遺伝子、XM_001817008で特定される遺伝子及びXM_001822481で特定される遺伝子については、被検物質の存在下における発現量が亢進していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質として同定することができる。また、XM_001820375で特定される遺伝子、XM_001727556で特定される遺伝子、XM_001727176で特定される遺伝子、XM_001727558で特定される遺伝子、XM_001727555で特定される遺伝子、XM_001820346で特定される遺伝子及びXM_001827554で特定される遺伝子については、被検物質の存在下における発現量が減少していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質として同定することができる。特に、細胞壁構築経路阻害剤の候補物質を同定する場合、表8に示した遺伝子のうちXM_001825115で特定される遺伝子の発現量を検討することが好ましい。XM_001825115で特定される遺伝子について被検物質の存在下における発現量が亢進していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質としての蓋然性が非常に高いと判断できる。
【0054】
さらにまた、Genbankアクセッション番号XM_658397で特定されるagsA遺伝子及び/又はGenbankアクセッション番号XM_655819で特定されるagsB遺伝子について、被検物質の存在下における発現量が亢進していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質として同定することができる。
【0055】
さらにまた、Genbankアクセッション番号XM_365841で特定されるcat1遺伝子について、被検物質の存在下における発現量が亢進していた場合には、当該被検物質をHOG経路活性化剤の候補物質として同定することができる。
【0056】
以上のように、種々の被検物質について、各種の抗菌作用を遺伝子発現パターンによって判定することで、抗菌剤の候補物質をスクリーニングすることができる。ただし、本スクリーニング方法においては、ある候補物質について特定の抗菌作用の有無を判定しても良いが、ある候補物質について、呼吸鎖阻害作用、エルゴステロール生合成阻害作用、細胞壁生合成阻害作用、MpkB経路阻害作用、細胞壁構築経路阻害作用及びHOG経路活性化作用のうち何れの抗菌作用を有するのかを判定しても良い。候補物質によっては、上述した各種抗菌作用のうち複数の抗菌作用を有するものとしてスクリーニングされる場合もある。
【0057】
スクリーニング方法2
次に、上述したスクリーニング方法2について説明する。スクリーニング方法2は、スクリーニング方法1とは異なり、上述したGenbankアクセッション番号で特定される遺伝子そのものの発現量を測定するのではなく、上述したGenbankアクセッション番号で特定される遺伝子のプロモータ領域を利用する方法である。詳細には、当該プロモータ領域と当該プロモータ領域の制御下に発現可能なレポーター遺伝子とを連結した組換えDNAを使用する。この組換えDNAはベクターに組み込まれて保存・使用することができる。ベクターとしては、何ら限定されないが、導入対象の微生物において複製可能なタイプのベクターを適宜選択して使用することが好ましい。
【0058】
上述したGenbankアクセッション番号で特定される遺伝子のプロモータ領域は、当該遺伝子の発現を制御するため、当該プロモータ領域はレポーター遺伝子についても同様に発現制御する。この組換えDNAを感染性微生物に導入した形質転換細胞に対して、候補物質を接触させ、被検物質の存在下におけるレポーター遺伝子の発現量と、被検物質の非存在下における発現量とを比較する。そして、公知の抗菌剤(呼吸鎖阻害剤、エルゴステロール生合成阻害剤、細胞壁生合成阻害剤、HOG経路活性化剤)を作用させた時と同様に又はmpkB遺伝子破壊株若しくはmpkA遺伝子破壊株と同様に、当該レポーター遺伝子の発現量が変動している場合、当該被検物質は、上述した抗菌作用を有するものとして同定することができる。
【0059】
ここで、プロモータ領域とは、上述したGenbankアクセッション番号で特定される遺伝子の開始コドンから上流の領域に存在し、転写因子が結合する領域を意味する。より具体的に、プロモータ領域は、上述したGenbankアクセッション番号で特定される遺伝子の開始コドンの上流2kbの領域、好ましくは上流1.5kbの領域に含まれている。さらに具体的に、配列番号1〜84に示した84個の遺伝子の開始コドンの上流2kbの領域の塩基配列を配列番号86〜169に示す。また、配列番号85に示した遺伝子の開始コドンの上流1.5kbの領域の塩基配列を配列番号170に示す。
【0060】
本スクリーニング方法において、プロモータ領域としては、配列番号86〜170に示した塩基配列からなるポリヌクレオチドを使用することができる。また、プロモータ領域としては、配列番号86〜170に示した塩基配列に対して70%以上、好ましくは80%以上、さらに好ましくは90%以上、より好ましくは95%以上、最も好ましくは98%以上の類似度(Similarity)を有する塩基配列からなるポリヌクレオチドを使用することができる。なお、類似度(Similarity)は、上述した定義と同様である。さらに、プロモータ領域としては、配列番号86〜170に示した塩基配列に対して相補的な塩基配列を有するポリヌクレオチドにストリンジェントな条件下でハイブリダイズするポリヌクレオチドを使用することができる。なお、ストリンジェントな条件は、上述した定義と同様である。
【0061】
ここで、レポーター遺伝子としては、いわゆるレポーターアッセイに使用されるものであれば特に限定されず、如何なる遺伝子を使用しても良い。レポーター遺伝子としては、例えば、CAT遺伝子、lacZ遺伝子、ルシフェラーゼ遺伝子、β-グルクロニダーゼ遺伝子(GUS)及びGFP遺伝子等を挙げることができる。レポーター遺伝子は、上述したプロモータ領域の制御下に発現可能なように当該プロモータ領域と連結される。換言すると、プロモータ領域に転写因子が結合することにより、レポーター遺伝子の発現が誘導されるように、プロモータ領域とレポーター遺伝子とを結合する。
【0062】
レポーター遺伝子の発現レベルは、使用するレポーター遺伝子の種類に応じて、当業者に公知の方法により測定することができる。例えば、レポーター遺伝子がCAT遺伝子である場合には、該遺伝子産物によるクロラムフェニコールのアセチル化を検出することによって、レポーター遺伝子の発現レベルを測定することができる。レポーター遺伝子がlacZ遺伝子である場合には、該遺伝子発現産物の触媒作用による色素化合物の発色を検出することにより、また、ルシフェラーゼ遺伝子である場合には、該遺伝子発現産物の触媒作用による蛍光化合物の蛍光を検出することにより、また、β-グルクロニダーゼ遺伝子(GUS)である場合には、該遺伝子発現産物の触媒作用によるGlucuron(ICN社)の発光や5-ブロモ-4-クロロ-3-インドリル-β-グルクロニド(X-Gluc)の発色を検出することにより、さらに、GFP遺伝子である場合には、GFPタンパク質による蛍光を検出することにより、レポーター遺伝子の発現レベルを測定することができる。
【0063】
本スクリーニング方法においても、上述したスクリーニング方法1と同様に、例えば、表5に示したGenbankアクセッション番号によって特定される遺伝子のうち少なくとも1以上、好ましくは5以上、より好ましくは10以上の遺伝子のプロモータ領域を使用し、被検物質の存在下におけるレポーター遺伝子の発現量が亢進していた場合には、当該被検物質を呼吸鎖阻害剤の候補物質として同定することができる。特に、呼吸鎖阻害剤の候補物質を同定する場合、表5に示した遺伝子のうちXM_001403469で特定される遺伝子のプロモータ領域を用いてレポーター遺伝子の発現量を検討することが好ましい。XM_001403469で特定される遺伝子のプロモータ領域を使用した場合に、被検物質の存在下における発現量が亢進していた場合には、当該被検物質を呼吸鎖阻害剤の候補物質としての蓋然性が非常に高いと判断できる。
【0064】
また、例えば、表6に示したGenbankアクセッション番号によって特定される遺伝子のうち少なくとも1以上、好ましくは3以上、より好ましくは5以上の遺伝子のプロモータ領域を使用し、被検物質の存在下におけるレポーター遺伝子の発現量が亢進していた場合には、当該被検物質をエルゴステロール生合成阻害剤の候補物質として同定することができる。特に、エルゴステロール生合成阻害剤の候補物質を同定する場合、表6に示した遺伝子のうちXM_362183で特定される遺伝子のプロモータ領域を用いてレポーター遺伝子の発現量を検討することが好ましい。XM_362183で特定される遺伝子のプロモータ領域を使用した場合に、被検物質の存在下における発現量が亢進していた場合には、当該被検物質をエルゴステロール生合成阻害剤の候補物質としての蓋然性が非常に高いと判断できる。
【0065】
さらに、例えば、表7に示したGenbankアクセッション番号によって特定される遺伝子のうち少なくとも1以上、好ましくは5以上、より好ましくは10以上、更に好ましくは20以上の遺伝子のプロモータ領域を使用し、被検物質の存在下におけるレポーター遺伝子の発現量が亢進していた場合には、当該被検物質を細胞壁生合成阻害剤の候補物質として同定することができる。特に、細胞壁生合成阻害剤の候補物質を同定する場合、表7に示した遺伝子のうちXM_364794で特定される遺伝子のプロモータ領域及び/又はXM_368552で特定される遺伝子のプロモータ領域を用いてレポーター遺伝子の発現量を検討することが好ましい。XM_364794で特定される遺伝子のプロモータ領域又はXM_368552で特定される遺伝子のプロモータ領域を使用した場合に、被検物質の存在下における発現量が亢進していた場合には、当該被検物質を細胞壁生合成阻害剤の候補物質としての蓋然性が非常に高いと判断できる。
【0066】
さらにまた、例えば、Genbankアクセッション番号XM_653297で特定されるスーパーオキシドデスムターゼ遺伝子のプロモータ領域を使用し、被検物質の存在下におけるレポーター遺伝子の発現量が亢進していた場合には、当該被検物質をMpkB経路阻害剤の候補物質として同定することができる。
【0067】
さらにまた、例えば、表8に示したGenbankアクセッション番号によって特定される遺伝子のうち少なくとも1以上、好ましくは3以上、より好ましくは5以上の遺伝子のプロモータ領域を使用し、被検物質の存在下におけるレポーター遺伝子の発現量が変動していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質として同定することができる。より具体的に、XM_001825115で特定される遺伝子のプロモータ領域、XM_001820488で特定される遺伝子のプロモータ領域、XM_001817008で特定される遺伝子のプロモータ領域及びXM_001822481で特定される遺伝子のプロモータ領域については、被検物質の存在下におけるレポーター遺伝子の発現量が亢進していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質として同定することができる。また、XM_001820375で特定される遺伝子のプロモータ領域、XM_001727556で特定される遺伝子のプロモータ領域、XM_001727176で特定される遺伝子のプロモータ領域、XM_001727558で特定される遺伝子のプロモータ領域、XM_001727555で特定される遺伝子のプロモータ領域、XM_001820346で特定される遺伝子のプロモータ領域及びXM_001827554で特定される遺伝子のプロモータ領域については、被検物質の存在下におけるレポーター遺伝子の発現量が減少していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質として同定することができる。特に、細胞壁構築経路阻害剤の候補物質を同定する場合、表8に示した遺伝子のうちXM_001825115で特定される遺伝子のプロモータ領域を使用してレポーター遺伝子の発現量を検討することが好ましい。XM_001825115で特定される遺伝子のプロモータ領域を使用して被検物質の存在下におけるレポーター遺伝子の発現量が亢進していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質としての蓋然性が非常に高いと判断できる。
【0068】
さらにまた、Genbankアクセッション番号XM_658397で特定されるagsA遺伝子のプロモータ領域及び/又はGenbankアクセッション番号XM_655819で特定されるagsB遺伝子のプロモータ領域を使用して、被検物質の存在下におけるレポーター遺伝子の発現量が亢進していた場合には、当該被検物質を細胞壁構築経路阻害剤の候補物質として同定することができる。
【0069】
さらにまた、Genbankアクセッション番号XM_365841で特定されるcat1遺伝子のプロモータ領域について、被検物質の存在下におけるレポーター遺伝子の発現量が亢進していた場合には、当該被検物質をHOG経路活性化剤の候補物質として同定することができる。
【0070】
以上のように、種々の被検物質について、各種の抗菌作用をレポーター遺伝子の発現パターンによって判定することで、抗菌剤の候補物質をスクリーニングすることができる。ただし、本スクリーニング方法においては、ある候補物質について特定の抗菌作用の有無を判定しても良いが、ある候補物質について、呼吸鎖阻害作用、エルゴステロール生合成阻害作用、細胞壁生合成阻害作用、MpkB経路阻害作用、細胞壁構築経路阻害作用及びHOG経路活性化作用のうち何れの抗菌作用を有するのかを判定しても良い。候補物質によっては、上述した各種抗菌作用のうち複数の抗菌作用を有するものとしてスクリーニングされる場合もある。
【実施例】
【0071】
以下、実施例を用いて本発明をより詳細に説明するが、本発明の技術的範囲は以下の実施例に限定されるものではない。
〔実施例1〕
呼吸鎖阻害剤検出マーカーの選抜方法
呼吸鎖にその作用を示す薬剤を検出することのできるマーカー遺伝子の選抜方法を以下に示す。
【0072】
呼吸鎖阻害剤暴露状態でのマイクロアレイによる発現解析
(1)トータルRNA抽出
トータルRNA抽出に用いた菌体は、100mlのYG液体培地を入れた300ml容三角フラスコにマグナポルテ・グリセア野生株Guy11株の分生子を約2×106個分生子になるように接種し、26℃、48時間緩やかに振とう培養後、呼吸鎖Complex1阻害剤としてジフルメトリム及びテブフェンピラド、Complex2阻害剤としてメプロニル、Complex3阻害剤としてクレソキシムメチル及びピリベンカルブ、Complex5(F1Fo ATPase)阻害剤としてオリゴマイシン、電子伝達に作用してATP合成を阻害する脱共役作用剤としてフルアジナムを0.01〜30ppmの濃度添加し、さらに26℃で2時間培養した。本薬剤処理を行った菌体及び無処理菌体は、ミラクロス(CALBIOCHEM社製)で集菌した。この回収した菌を乳鉢中で液体窒素を注ぎながらパウダー状になるまで破砕した。パウダー状の菌体はRNA Plant Mini Kit(キアゲン社製)を用いて、添付マニュアルに従いトータルRNA抽出を行った。
【0073】
(2)マイクロアレイ
マイクロアレイに用いるDNAチップはマグナポルテ・グリセアの10,000遺伝子から設計した60merのオリゴプローブをインクジェット方式でスポットした独自のマイクロアレイを使用し、抽出した薬剤処理区をtestサンプルに、薬剤無処理区を比較対象にCyanine3及びCyanine5を用いた二色法により行った。一回の実験区ごとにCyanine3とCyanine5のカラースワップを行う事により計2回の反復実験を行った。
【0074】
(3)RNAの標識(Cyanine5、Cyanine3)
RNAのCyanine3及びCyanine5標識はmRNAを用いて行い、トータルRNAからのmRNAの精製はOligotex-dT30<super> mRNA purification Kit(タカラバイオ社製)を用い、本キットに添付のマニュアルに従った。mRNAからのcDNAの作製及び標識はCyScribe cDNA Post Labelling Kit(GE helthcare)を用い、本キットに添付のマニュアルに従い行った。
【0075】
(4)ハイブリダイゼーション
Cyanine3及びCyanine5標識済みcDNA混合サンプルに、表9に示したハイブリダイゼーションBufferを60μl加え溶解後、95℃で5分熱変性を行った。その後30分間室温で保持した後、DNAチップ上に滴下し、カバーガラスをのせた。カバーガラスをのせたDNAチップは温水上に浮かべたhumidity chamber上に置き42℃、一晩インキュベーションすることによりハイブリダイゼーションを行った。
【0076】
【表9】
【0077】
(5)ポストハイブリダイゼーション
42℃で一晩インキュベーション後、DNAチップは表10に示したWash buffer1中でカバーガラスをはずした後、さらに新しいWash Buffer1で15分、次いで表11に示したWash Buffer2で5分、表12に示したWash Buffer3で5分の順に振とうすることにより洗浄を行った。その後、さらにdH2O中で2分間洗浄後800×g、2分の遠心により水分を飛ばしGenePix 4000B(Axon instrument)を用いてスポットのスキャンを行った。
【0078】
【表10】
【0079】
【表11】
【0080】
【表12】
【0081】
マイクロアレイの結果から抽出された呼吸鎖阻害剤処理により共通して発現する遺伝子のうち、呼吸鎖阻害剤及びその他抗菌剤の薬物代謝に関わる遺伝子を除いた遺伝子を呼吸鎖阻害剤の検出マーカー候補とした。以下の表13にその上位10遺伝子のマイクロアレイ解析結果を表示する。値は無処理区と比較した場合の相対値で示し、発現比1.5倍以上の値を網掛けで表示した。
【0082】
【表13】
【0083】
〔実施例2〕
呼吸鎖阻害剤検出マーカー遺伝子の検証
表13に示した遺伝子が呼吸鎖阻害剤を処理したときに、特異的に発現変化があり検出マーカーとして利用可能か否かをMGG_12936シアン耐性呼吸酵素遺伝子(AOX)及びMGG_04999NADH-デヒドロゲナーゼ遺伝子を例に検証した。
【0084】
(1)トータルRNA抽出
トータルRNA抽出に用いた菌体は、100mlのYG液体培地を入れた300ml容三角フラスコにマグナポルテ・グリセア野生株Guy11株の分生子を約2×106個分生子になるように接種し26℃、48時間緩やかに振とう培養。その後、指定の薬剤を各濃度添加し、さらに26℃で30分、2時間及び6時間培養した。薬剤処理に用いた呼吸鎖阻害薬剤及び処理濃度は呼吸鎖Complex1阻害剤としてジフルメトリム、Complex2阻害剤としてメプロニル、Complex3阻害剤としてクレソキシムメチル、Comlex5(F1Fo ATPase)阻害剤としてオリゴマイシン、電子伝達に作用してATP合成を阻害する脱共役作用剤としてフルアジナムをそれぞれ、1ppm、30ppm、20ppm、0.5ppm、0.01ppmとした。その他、対照薬剤として、フラバノン系化合物サクラネチン10ppm、MAPキナーゼシグナリングに作用すると考えられるフルジオキソニル20ppm、プロシミドン10ppm、イプロジオン30ppm、エルゴステロール生合成阻害剤オキスポコナゾール・フマル酸塩1ppmを用いた。本薬剤処理により得た菌体及び無処理菌体は、ミラクロス(CALBIOCHEM社製)で集菌した。回収した菌を乳鉢中で液体窒素を注ぎながらパウダー状になるまで破砕した。パウダー状の菌体はRNA Plant Mini Kit(キアゲン社製)を用いて、添付マニュアルに従いトータルRNA抽出を行った。
【0085】
(2)cDNAの合成
cDNAの合成には、トータルRNAを用いた。260 nmにおける吸光度からの計算値で5μg量のトータルRNAをRNase freeの1.5mlチューブに移し、さらにDEPC処理水を加え100μlとした。これにRNase free DNase Iを5unit加え、37℃で1時間反応させ混在するゲノムDNAの分解を行った。その後、ExScript RT reagent Kit(タカラバイオ社製)を用いて添付マニュアルに従って逆転写反応を行いcDNAを得た。
【0086】
(3)定量RT-PCR
合成したcDNAの蛍光色素によるラベリング反応には、DyNAmo SYBR Green qPCR Kit (Finnzymes社製)を用いた。スキャナー解析はDNA Engine OPTICON2(MJ Research社製)を、データ解析には解析ソフトOpticon Monitor2 ver.2.02(MJ Research社製)を用いた。反応操作は、DyNAmo SYBR Green qPCR Kit添付マニュアルにしたがった。反応条件は95℃、10分間のヒートショック後、熱変性95℃、10秒、アニーリング60℃、30秒、伸長反応72℃、30秒、プレートリードの反応を40 サイクル行い、60〜95℃のグラジェント(0.2℃おきに1秒間hold)でDissociation Curveの作成を行った。PCRは各cDNA、各標的遺伝子につき三連で行った。
【0087】
呼吸鎖阻害剤処理cDNAサンプルを用いてシアン耐性呼吸酵素遺伝子(AOX)及びNADH-デヒドロゲナーゼ遺伝子の発現を経時的に調べた。またコントロールとしてActinタンパク質をコードしていると推定される遺伝子の転写量を調べた。それぞれ以下の特異的プライマーを用いた。
Actin-F:ATGCCATCGGAAAGACAGAC(配列番号171)
Actin-R:CAGGAGTCGATCTCCAAAGC(配列番号172)
AOX_RTF:TTGTCGCATACCTCATCAGC(配列番号173)
AOX_RTR:CAATTTCTGGCACCTGGAAC(配列番号174)
NADH_RTF:AATCACCAAGGATGGACGAC(配列番号175)
NADH_RTR:CCTAGGTATGCCATGGTTCC(配列番号176)
【0088】
PCR終了後、専用の解析ソフトOpticon Monitor2 ver.2.02(MJ Research社)を用いて解析を行った。Quantitation画面上で各遺伝子のPCR産物が対数的に増幅し始めるサイクル数(CT)を決定した。この値をもとに各cDNAの標的遺伝子の相対発現量を以下の計算方法から求めた。
a) ΔCT値の算出(各cDNAの標的遺伝子とγ-actinとの差)
ΔCT = (y-x)
ここでxはCT (γ-actin)であり、yはCT(Target gene)である
b) ΔCT値の算出(あるcDNAのΔCTと基準とするcDNAのΔCTとの差)
ΔCT =ΔCT (C) -ΔCT (S)
ここで、CはComparative cDNAであり、SはStandard cDNAである
c) 相対発現量の算出
2-(ΔCT)
【0089】
RT-PCRの結果をシアン耐性呼吸酵素遺伝子(AOX)については図1の上段に、NADH-デヒドロゲナーゼ遺伝子については図1の下段にactin当たりの発現量をグラフ化したものを示した。野生株を呼吸鎖阻害剤に曝すと、シアン耐性呼吸酵素遺伝子(AOX)及びNADH-デヒドロゲナーゼ遺伝子の転写量は増加していた。一方、呼吸鎖への作用機作を有しない化合物に関しては、それぞれの遺伝子発現の顕著な増加は認められなかった。このことにより、今回例として設定したシアン耐性呼吸酵素遺伝子(AOX)及びNADH-デヒドロゲナーゼ遺伝子は呼吸鎖に作用を及ぼす薬剤特異的に発現が増加していると考えられる。よって、例えば、シアン耐性呼吸酵素遺伝子 (AOX)及びNADH-デヒドロゲナーゼ遺伝子の5'-UTR(5'上流領域のプロモータ領域)はレポーター遺伝子と機能的に結合させることにより、呼吸鎖阻害剤のスクリーニング系として利用できる可能性が確認された。
【0090】
〔実施例3〕
呼吸鎖阻害剤検出レポーター
実施例2で呼吸鎖阻害剤を暴露した時に特異的な遺伝子の発現増を示すシアン耐性呼吸酵素遺伝子(AOX)を例にして、本遺伝子の5'-UTR(5'上流領域のプロモータ)をレポーター遺伝子と機能的に結合させることによって転写量の呼吸鎖特異的な増加を可視化するシステムを構築した例を示す。
【0091】
(1)シアン耐性呼吸酵素遺伝子(AOX) 5'領域のクローニング
Broad Institute のマグナポルテ・グリセア(Magnaporthe grisea)ゲノムデータベース(http://www.broad.mit.edu/annotation/genome/magnaporthe_grisea/MultiHome.html)からAOX 遺伝子の5'領域の塩基配列情報を取得し、その配列情報を参考に一組のオリゴヌクレオチドを設計した(5'-CCAAGCTTCCTTCCTCACTA-3'(配列番号177)、5'-CCAAGCTTAAGACGGTACGTTCAGTGT-3'(配列番号178))。マグナポルテ・グリセアGuy11株のゲノムDNAをテンプレートとしてPCR反応を行った。増幅反応は、94℃、3分間鋳型DNAを変性し、94℃、30秒間、55℃、30秒間、72℃、2分間保持するサイクルを35サイクルおこなった後、72℃、5分間で完全伸長させ、4℃で保持した。PCR用装置は、PCR Thermal Cycler PERSONAL (タカラバイオ社製)を用いた。このPCRによる増幅断片をアガロース電気泳動にて確認を行ったところ約889塩基対の増幅が見られた。この増幅断片をアガロースゲル電気泳動に供したゲル中よりGENECLEAN IIIキット(Qbiogene社製)を用いてDNAを抽出し、これを挿入DNA断片とした。この挿入DNA断片をpGEM-T Easy Vector System(Promega社製)を用いてpGEM-T Easy Vectorに連結させ、連結DNA溶液を得た。連結DNA溶液10μlを氷中でよく冷却した後、氷上解凍したコンピテントセルJM109を100μl加え穏やかに撹拌し、氷中で20分間、続いて42℃で45秒ヒートショック処理を行った。これに、400μlのLB液体培地を加え、37℃で1時間培養後、100μg/mlのアンピシリンを添加したLB平板培地にまき、37℃で一晩培養した。目的のプラスミドDNAを用いて形質転換した大腸菌の単一のコロニーを3mlの100μg/mlのアンピシリンを添加したLB液体培地に植菌し、37℃で一晩振盪培養した。1.5mlの培養液を1.5mlのエッペンドルフチューブに移して15,000×gで1分間遠心分離し、沈殿を100μlの氷冷したTEG(25mM Tris-HCl、10mM EDTA、50mM Glucose、pH8.0)で懸濁し、これに200μlの0.2N NaOH-1%SDSを加えて穏やかに撹拌した後、150μlの3M NaOAc(pH5.2)を加えて混合した。これを15,000×g、4℃で5分間遠心分離し上清を回収し450μlのフェノール・クロロホルム・イソアミルアルコール(25:24:1)を加えて激しく撹拌した後、15,000×g、室温で5分間遠心分離し上層を回収した。この溶液に-20℃で氷冷した900μlのエタノールを加えて-20℃で10分間放置後、4℃にて15,000×gで5分間遠心分離した。沈殿を500μlの70%エタノールでリンスしたのち、乾燥させ、最後にRNase(100μg/ml)を含むTE(10mM Tris-HCl、1mM EDTA、pH8.0)50μlに溶解した。得られたプラスミドをHindIIIで切断後、アガロース電気泳動により挿入断片の存在を確認し、このプラスミド中の挿入DNA断片を添付のプロトコールに従い、ABI PRISMTM 377 DNA sequencing system (PE Biosystem社製)にて解析した。889塩基対からなるシアン耐性呼吸酵素遺伝子(AOX)5'上流領域のプロモータの塩基配列を配列番号179に示す。また、本ベクターをpGEM-PrAOXとする。
【0092】
(2)EGFPベクターの作成
pCAMBIA-0380バイナリーベクター(CAMBIAより購入)を骨格にし、薬剤選択マーカーとしてハイグロマイシンB耐性遺伝子Hphを融合することにより作成した糸状菌形質転換ベクターpCH11(図2)をHindIII及びBglIIで消化し、そこにEGFP-LacI-TagdA (p3'SS d EGFPと大腸菌LacI:Lactose operon transcriptional repressor Lac1(E. coli EU337980, 4-1068bp)及びAspergillus oryzae Α-グルコシダーゼ遺伝子agdA ターミネーター(Aspergillus oryzae、Supercontig 6: 3289259-3289838 +)融合体)のHindIII-BglII断片を組み合すことによりEGFPベクター、pCH11-EGFP-LacIを作成した(図3)。
【0093】
(3)AOX-EGFP-LacIレポーター(レポーター遺伝子ベクター)の作製
pCH11-EGFP-LacI(図2)プラスミドをHindIIIで消化し、そこにpGEM-PrAOXをHindIII処理することにより得たシアン耐性呼吸酵素遺伝子(AOX)5'領域を定法により融合した。本レポーターアッセイプラスミドであるAOX-EGFP-LacIレポーターベクターをpCH11-PrAOX-EGFP-LacIと命名した。
【0094】
(4) pCH11-PrAOX-EGFP-LacIプラスミドを用いたマグナポルテ・グリセアの形質転換
マグナポルテ・グリセアの形質転換はアグロバクテリウム法(Gento Tsuji,Satoshi Fujii, Naoki Fujihara, Chika Hirose,Seiji Tsuge, Tomonori Shiraishi and Yasuyuki Kubo(2003):Agrobacterium tumefaciens-mediated transformation for random insertional mutagenesis in Colletorichum lagenarium. Journal of General Plant Pathology 69:230-239)を改良した方法を用いた。得られた菌株へのベクター挿入の確認は、PCR、シークエンス及びサザン解析によって行った。
【0095】
(5)レポーター作動の確認
レポーター作動の確認は、呼吸鎖阻害剤であるジフルメトリム、クレソキシムメチル、オリゴマイシン及び比較薬剤としてオキスポコナゾール、フェンヘキサミド、カルプロパミド、ベノミルで菌体を処理した時のEGFPの蛍光を観察することによって行った。マグナポルテ・グリセアPAOX-NLS株形質転換株の分生子を2×106個分生子/100mlになるように、100mlのYG液体培地を入れた300ml容坂口フラスコに接種し次の条件で培養した。
(条件1) 26℃、24時間培養
(条件2) 26℃、24時間培養した後、上記薬剤を添加し、さらに26℃で6時間培養。
その後、各処理菌株の蛍光を蛍光顕微鏡を用いて観察した。
【0096】
(6)EGFP蛍光観察
スライドガラスに培養後の菌糸体をのせGFPの蛍光観察を行った。各種薬剤添加6時間後、呼吸鎖阻害剤(ジフルメトリム、クレソキシムメチル、オリゴマイシン)処理においてのみ、核に局在した強いEGFP-LacI蛍光が観察され、無処理区及び呼吸鎖阻害剤以外の化合物処理ではEGFP-LacI蛍光はほとんど観察されなかった(図4)。以上の結果から、pAOX-EGFP-LacIレポーターは呼吸鎖阻害剤の特異的スクリーニングに利用できると言える。
【0097】
〔実施例4〕
エルゴステロール生合成阻害剤検出マーカーの選抜方法
エルゴステロールの生合成にその作用を示す薬剤を検出することのできるマーカー遺伝子の選抜方法は、呼吸鎖阻害剤の代わりにエルゴステロール生合成阻害剤であるオキスポコナゾール・フマル酸塩を1及び5ppm、フェンヘキサミドを0.5ppmの濃度で使用した以外は実施例1と同様に行った。
【0098】
マイクロアレイの結果から抽出されたエルゴステロール生合成阻害剤で共通して発現する遺伝子のうち、エルゴステロール生合成阻害剤及びその他抗菌剤の薬物代謝に関わる遺伝子を除いた遺伝子をエルゴステロール生合成阻害剤の検出マーカー候補とした。以下の表14にその上位5遺伝子のマイクロアレイ解析の結果を表示する。値は無処理区と比較した場合の相対値で示し、発現比1.5倍以上の値を網掛けで表示した。
【0099】
【表14】
【0100】
〔実施例5〕
エルゴステロール生合成阻害剤検出マーカー遺伝子の検証
表14に示した遺伝子がエルゴステロール生合成阻害剤を処理したときに、特異的に発現変化があり検出マーカーとして利用可能か否か、Cytochrome P450ステロール14αジメチラーゼ(MGG_04628)及びステロール24Cメチルトランスフェラーゼ(MGG_10860)を例に検証した。
【0101】
本実施例では、Cytochrome P450ステロール14αジメチラーゼ(MGG_04628)及びステロール24Cメチルトランスフェラーゼ(MGG_10860)を特に検出するため、以下の特異的プライマーを使用した以外は実施例2と同様にして遺伝子の転写量を調べた。
SteCyp51-F:CCACATGATGATCACGCTTC(配列番号180)
SteCyp51-R:GTCTCCTTGACCACGTTTGC(配列番号181)
24C-F:AACATGCAGACAGCCAAGAC(配列番号182)
24C-R:ATTCGAAATGTTCCCAGCAC(配列番号183)
【0102】
RT-PCRの結果をCytochrome P450ステロール14αジメチラーゼ(MGG_04628)については図5の上段に、ステロール24Cメチルトランスフェラーゼ(MGG_10860)については図5の下段にactin 当たりの発現量をグラフ化したものを示した。野生株を各種薬剤に曝すと、Cytochrome P450ステロール14αジメチラーゼ(MGG_04628)及びステロール24Cメチルトランスフェラーゼ(MGG_10860)の転写量はエルゴステロール生合成阻害剤オキスポコナゾール添加後増加していた。このことにより、今回例として設定したCytochromeP450ステロール14αジメチラーゼ(MGG_04628)及びステロール24Cメチルトランスフェラーゼ(MGG_10860)はエルゴステロール生合成に作用を及ぼす薬剤処理下で発現が増加していると考えられる。よって、例えば、Cytochrome P450ステロール14αジメチラーゼ(MGG_04628)及びステロール24Cメチルトランスフェラーゼ(MGG_10860)の5'-UTR(5'上流領域のプロモーター)は、レポーター遺伝子と機能的に結合させることにより、エルゴステロール生合成阻害剤のスクリーニング系として利用できる可能性が確認された。
【0103】
〔実施例6〕
エルゴステロール生合成阻害剤検出レポーター
実施例5でエルゴステロール生合成阻害剤を暴露した時に、特異的な遺伝子の発現増を示すCytochromeP450ステロール14αジメチラーゼ(MGG_04628)を例にして、本遺伝子の5'-UTR(5'上流領域のプロモーター)をレポーター遺伝子と機能的に結合させることによって転写量のエルゴステロール生合成阻害剤特異的な増加を可視化するシステムを構築した例を示す。
【0104】
Broad Instituteのマグナポルテ・グリセア(Magnaporthe grisea)ゲノムデータベース (http://www.broad.mit.edu/annotation/genome/magnaporthe_grisea/MultiHome.html)からCytochrome P450ステロール14αジメチラーゼ(MGG_04628)の5'領域の塩基配列情報を取得し、その配列情報を参考に一組のオリゴヌクレオチドを設計した(5'-CCAAGCTTATGTGAGCCATCTTCGGATT-3'(配列番号184)、5'-CCAAGCTTGACGATCGGGTAAGGAGACT-3'(配列番号185))。マグナポルテ・グリセアGuy11株のゲノムDNAをテンプレートとしてPCR反応を行った。増幅反応は、94℃、3分間鋳型DNAを変性し、94℃、30秒間、55℃、30秒間、72℃、2分間保持するサイクルを35サイクルおこなった後、72℃、5分間で完全伸長させ、4℃で保持した。PCR用装置はPCR Thermal Cycler PERSONAL(タカラバイオ社製)を用いた。このPCRによる増幅断片をアガロース電気泳動にて確認を行ったところ約1,405塩基対の増幅が見られた。
【0105】
その後、実施例3と同様にしてプロモータ領域の塩基配列を解析した結果を配列番号186に示す。また、本ベクターをpGEM-PrCyp51とする。
【0106】
また、実施例3と同様にして、Cyp51-EGFP-LacIレポーターを作製し、レポーター作動を確認した。その結果を図6に示した。各種薬剤添加6時間後、オキスポコナゾール・フマル酸塩の処理においてのみ、核に局在した強いEGFP-LacI蛍光が観察され、それ以外の化合物処理ではEGFP-LacI蛍光はほとんど観察されなかった。以上の結果から、pCyp51-EGFP-LacIレポーターシステムはエルゴステロール生合成阻害剤のスクリーニングに利用できると言える。
【0107】
〔実施例7〕
細胞壁生合成阻害剤の検出マーカーの選抜方法
細胞壁の生合成にその作用を示す薬剤を検出することのできるマーカー遺伝子の選抜方法は、呼吸鎖阻害剤の代わりに細胞壁生合成阻害剤である1μgミカファンギン(アステラス製薬)及び1mgカルコフロール・ホワイト(シグマ・アルドリッチ)を使用した以外は実施例1と同様に行った。
【0108】
マイクロアレイの結果から抽出された細胞壁生合成阻害剤により共通に発現が誘導される、もしくは抑制される遺伝子のうち、細胞壁生合成阻害剤の薬物代謝に関わる遺伝子を除いた遺伝子を細胞壁生合成阻害剤の検出マーカー候補とした。以下の表15にそのマイクロアレイ結果の一部を表示する。値は無処理区と比較した場合の相対値で示した。
【0109】
【表15】
【0110】
〔実施例8〕
細胞壁生合成阻害剤検出レポーター
表15に示した遺伝子が細胞壁生合成阻害剤を処理したときに、特異的に発現変化があり検出マーカーとして利用可能か否か検証した。本実施例では、以下の特異的プライマーを使用した以外は実施例2と同様にして遺伝子の転写量を調べた。
MGG_02004-F:GGACAAAATGACAGTCCA(配列番号187)
MGG_02004-R:CCAAGTTTTCTTTCGGTTTC(配列番号188)
MGG_00692-F:TACAGACATGCCGCCGGGTGGACAA(配列番号189)
MGG_00692-R:ACGCTTGAGGTCCATCTGACCACTGCT(配列番号190)
MGG_04943-F:CCTTCTCGATCGCATGCT(配列番号191)
MGG_04943-R:ATCTGACCGACGTACTCTTG(配列番号192)
MGG_09639-F:TGTTTTCGTCGCAGTTCGTC(配列番号193)
MGG_09639-R:GCTCCTTTCTAAAGACCTTGGA(配列番号194)
MGG_05133-F:GTTGTTTCTGAGACCGGCCTGGTCTAC(配列番号195)
MGG_05133-R:CCATCGCTGATGGGTCCATGCCTACA(配列番号196)
MGG_02773-F:GATCATGGAACGGCAAAG(配列番号197)
MGG_02773-R:GTACCCCATTTCACCAAAC(配列番号198)
【0111】
RT-PCRの結果を図7にactin当たりの発現量をグラフ化したものを示した。野生株を細胞壁ストレス剤に曝すと、マイクロアレイの結果と同様に、4遺伝子(MGG_02004、MGG_00692、MGG_04943及びMGG_09639)の転写量は細胞壁生合成阻害剤添加後増加し、2遺伝子(MGG_05133及びMGG_02773)の転写量は減少していた。このことにより、今回例として設定した上記6遺伝子は細胞壁生合成に作用を及ぼす薬剤特異的に発現が変動していると考えられる。よって、例えば、これらの5'-UTR(5'上流領域のプロモータ)はレポーター遺伝子と機能的に結合させることによって、本発明のスクリーニング方法においてレポーター遺伝子の転写量の変化を検出することにより、細胞壁生合成阻害剤のスクリーニング系として利用できる可能性が確認された。
【0112】
〔実施例9〕
細胞壁生合成阻害剤のスクリーニングに用いるレポーターアッセイ株の作出
実施例8で細胞壁生合成阻害剤を曝露した時に、特異的な遺伝子の発現増を示す2遺伝子(MGG_00692及びMGG_09639)を例にして、これら遺伝子の5'-UTR(5'上流領域のプロモータ)をレポーター遺伝子と機能的に結合させることによって転写量の細胞壁生合成阻害剤特異的な増加を可視化するシステムを構築した例を示す。
【0113】
(1)遺伝子(MGG_00692) 5'領域のクローニング
Broad Instituteのマグナポルテ・グリセア(Magnaporthe grisea)ゲノムデータベース(http://www.broad.mit.edu/annotation/genome/magnaporthe_grisea/MultiHome.html)から遺伝子(MGG_00692)の5'領域の塩基配列情報を取得し、その配列情報を参考に一組のオリゴヌクレオチドを設計した(5'-CTTAGGCAACCTTGACGGCC-3'(配列番号199)、5'-GTCGTGTCCTTGGTTCATGG-3'(配列番号200))。マグナポルテ・グリセアGuy11株のゲノムDNAをテンプレートとしてPCR反応を行った。増幅反応は、94℃、3分間鋳型DNAを変性し、94℃、30 秒間、55℃、30秒間、72℃、2分間保持するサイクルを35サイクルおこなった後、72℃、5分間で完全伸長させ、4℃で保持した。PCR用装置はPCR Thermal Cycler PERSONAL(タカラバイオ社製)を用いた。このPCRによる増幅断片をアガロース電気泳動にて確認を行ったところ約1,000塩基対の増幅が見られた。
【0114】
その後、実施例3と同様にしてプロモータ領域の塩基配列を解析した結果を配列番号201に示す。また、本ベクターをpGEM-MSTU1とする。
【0115】
(2)遺伝子(MGG_09639)5'領域のクローニング
Broad Instituteのマグナポルテ・グリセア(Magnaporthe grisea)ゲノムデータベース (http://www.broad.mit.edu/annotation/genome/magnaporthe_grisea/MultiHome.html)から遺伝子(MGG_09639)の5'領域の塩基配列情報を取得し、その配列情報を参考に一組のオリゴヌクレオチドを設計した(5'-GTCCCGACAGCCAGTGTGCA-3'(配列番号202)、5'-GTTTTTTTACGATACCACCATAAAGTG-3'(配列番号203))。マグナポルテ・グリセアGuy11株のゲノムDNAをテンプレートとしてPCR反応を行った。増幅反応は、94℃、3分間鋳型DNAを変性し、94℃、30 秒間、55℃、30秒間、72℃、2分間保持するサイクルを35サイクルおこなった後、72℃、5分間で完全伸長させ、4℃で保持した。PCR用装置は PCR Thermal Cycler PERSONAL(タカラバイオ社製)を用いた。このPCRによる増幅断片をアガロース電気泳動にて確認を行ったところ約1,000塩基対の増幅が見られた。
【0116】
その後、実施例3と同様にしてプロモータ領域の塩基配列を解析した結果を配列番号204に示す。また、本ベクターをpGEM-AGS1とする。
【0117】
(3)EGFPベクターの作成
Aspergillus nidulans用シャトルベクター pAUR316(タカラバイオ社製)の自律複製AMA配列を除いたpNA316のBgl IIサイトに、EGFP-LacI-NLS-TagdA (p3'SS dEGFPと大腸菌LacI: Lactose operon transcriptional repressor Lac1(E. coli EU337980, 4-1068bp)及びSV40の核局在配列、Aspergillus oryzae Α-グルコシダーゼ遺伝子agdAターミネーター(Aspergillus oryzae、Supercontig 6: 3289259-3289838 +)融合体)を組み合わせたEGFPベクターpNA(N)EGFPを構築した(図8)。
【0118】
(4)pMSTU1-EGFPレポーター(レポーター遺伝子ベクター)の作製
pNA(N)EGFP(図8)プラスミドをNotIで消化し、そこにpGEM-MSTU1をNotI処理することにより得た 遺伝子MGG_00692の5'領域を定法により融合した。本レポーターアッセイプラスミドはpMSTU1-EGFPと命名した。
【0119】
(5)pAGS1-EGFPレポーター(レポーター遺伝子ベクター)の作製
pNA(N)EGFP(図8)プラスミドをNotIで消化し、そこにpGEM-AGS1をNotI処理することにより得た 遺伝子MGG_09639の5'領域を定法により融合した。本レポーターアッセイプラスミドはpAGS1-EGFPと命名した。
【0120】
(6)pMSTU1-EGFP及びpAGS1-EGFPを用いたマグナポルテ・グリセアの形質転換
マグナポルテ・グリセアの形質転換はPEG-プロトプラスト法(Leung, H., Lehtinen, U., Karjalainen, R., Skinner, D., Tooley, P., Leong, S., Ellingboe, A., 1990. Transformation of the rice blast fungus Magnaporthe grisea to hygromycin B resistance. Curr. Genet. 17, 409-411.)によって行った。得られた菌株へのベクター挿入の確認は、PCR、シークエンス及びサザン解析によって行った。
【0121】
(7)レポーター作動の確認
レポーター作動の確認は、実施例3と同様に行った。スライドガラスに培養後の菌糸体をのせGFPの蛍光観察を行った結果を図9に示す。細胞壁生合成阻害剤添加3時間後の処理において核に局在した強い緑色蛍光が観察され、無処理区では緑色蛍光はほとんど観察されなかった。以上の結果から、pMSTU1-EGFPレポーター及びpAGS1-EGFPレポーターは細胞壁生合成阻害剤の特異的スクリーニングに利用できる。
【0122】
〔実施例10〕
Aspergillus nidulans mpkB遺伝子破壊株の造成
(1)遺伝子欠失破壊用DNA断片の作製
アスペルギルス・オリゼ(Aspergillus oryzae)のargB遺伝子を欠失破壊株の選択用マーカーとして選択し、argB遺伝子を含むpAORBプラスミドから以下のように設計したプライマーを用いてargB遺伝子の断片をPCR増幅した(5'-GAATTCCATCCGCTGTGGCCGACTCA-3' (配列番号205)、5'-GGTACCGACGGAGTAACTGGAAAGATACGA-3' (配列番号206))。増幅にはTAKARAのPrimeStar HS DNA polymerase を用い、増幅反応は、95℃、2分間鋳型DNAを変性し、98℃、10秒間、55℃、5秒間、72℃、2分30秒間保持するサイクルを30サイクルおこなった後、72℃、5分間で完全伸長させ、4℃で保持した。このPCRによる増幅断片をアガロースゲル電気泳動にて確認を行ったところ約2700塩基対の増幅が見られた。
【0123】
次に、Broad Instituteのアスペルギルス属ゲノムデータベース(http://www.broad.mit.edu/annotation/genome/aspergillus_group/MultiHome.html)からアスペルギルス・ニドランス(Aspergillus nidulans)のmpkB遺伝子を含む上下流域の塩基配列を参考にプライマーを設計し、mpkB遺伝子の開始コドンから5'側上流領域(5'-UTR)と終止コドンから3'側下流領域(3'-UTR)の各1kbのDNA領域をPCRで増幅した。増幅にはタカラバイオ社製のPrimeStar HS DNA polymeraseを用い、増幅反応は、95℃、2分間鋳型DNAを変性し、98℃、10秒間、55℃、5秒間、72℃、2分間保持するサイクルを30サイクルおこなった後、72℃、5分間で完全伸長させ、4℃で保持した。このPCRによる増幅断片をアガロースゲル電気泳動にて確認を行ったところ約1,000塩基対の増幅が見られた。5'-UTR、前述のargB遺伝子断片、3'-UTRをfusion PCRにより連結する訳であるが、argB遺伝子断片との連結部になる位置にアニールするプライマーのテール部には、argB遺伝子断片の末端の配列を35 base導入した。各プライマーの配列を表16に記した。
【0124】
【表16】
【0125】
5'-UTR、argB遺伝子、3'-UTRの各断片をアガロースゲル電気泳動に供したゲル中よりillustra GFX PCR DNA and Gel Band Purification Kit(GE healthcare社製)を用いてDNAを抽出した。これら3断片を等量ずつ混合したDNA溶液をPCRの鋳型とし、fusion PCRを行った。fusion PCRは、両端に位置するANmpkB-5'UTR-FとANmpkB-3'UTR-Rのプライマーを供し、PrimeStar HS DNA polymerase(タカラバイオ社製)を用いて以下に記す増幅反応をおこなった。すなわち、94℃、2分間鋳型DNAを変性し、94℃、30秒間、58℃、45秒間、72℃、5分間保持するサイクルを30サイクルおこなった後、72℃、10分間で完全伸長させ、4℃で保持した。このPCRによる増幅断片をアガロースゲル電気泳動にて確認を行ったところ、3つの断片の合計サイズと同じ約4,700塩基対の増幅が見られた。この断片をアガロースゲル電気泳動に供したゲル中よりillustra GFX PCR DNA and Gel Band Purification Kit(GE healthcare社製)を用いてDNAを抽出し、mpkB遺伝子欠失破壊用DNA断片とした。
【0126】
(2)形質転換
mpkB遺伝子欠失破壊のための形質転換はプロトプラスト-PEG法を改良した方法を用い、形質転換するDNA断片は、上記で作製したmpkB遺伝子欠失破壊用DNA断片を用いた。アスペルギルス・ニドランスABPU1 ΔligD::ptrA(ビオチン(biA1)、アルギニン(argB2)、ウリジン(pyrG89)、ピリドキシン(pyroA4)要求性株(東北大学大学院農学研究科・藤岡智則博士より供与))の分生子懸濁液をYPD培地に植菌し、37℃、20時間振盪培養した。17G1滅菌済ガラスフィルターを用いて集菌後、菌体を50 ml容の遠心チューブに移し、滅菌水で洗浄した。その後、菌体を30 mlのプロトプラスト化溶液(0.8M NaCl、10mM NaH2PO4、10mg/ml Lysing enzyme (Sigma Chemical社製)、5mg/ml Cellulase Onozuka R-10 (Yakult Pharmaceutical Ind.社製)、2.5mg/ml Yatalase (タカラバイオ社製)を加え懸濁し、30℃、90rpm、3時間振盪しプロトプラスト化反応を行った。滅菌したMIRACLOTH(CALBIOCHEM社製)にて濾過し、濾液中のプロトプラストを3,000×g、4℃、5分間遠心分離して沈澱として得た。0.8M NaClにて1回プロトプラストを洗浄し、3,000×g、4℃、5分間遠心分離して沈澱させた。このプロトプラストをSolution 1(0.8M NaCl、10mM CaCl2、10mM Tris-HCl、pH8.0)で懸濁した。プロトプラスト懸濁液を200μlずつ15ml容の遠心チューブに移し、それぞれにSolution 2(40%(w/v) PEG♯4000、 50mM CaCl2、50mM Tris-HCl、pH8.0)40μlと前述の形質転換用DNA溶液各5μl(DNA量として5μg)を加えよく混合し、氷中で30分間放置した。1 mlのSol.2を加え混合し、室温で20分間放置した。5mlのSol.1で2回洗浄し、Sol.2をなるべく取り除いた。50℃に温めておいた終濃度0.02μg/mlのビオチン、5mMのウリジン、10mMのウラシル、0.5μg/mlのピリドキシンを添加したCzapek-Dox(CD)軟寒天培地にプロトプラスト縣濁液を加え混合し、終濃度0.02μg/mlのビオチン、5mMのウリジン、10mMのウラシル、0.5μg/mlのピリドキシンを添加したCD寒天培地に重層した。その後、30℃で分生子を形成するまで培養した。形質転換体からのmpkBΔ株の選択は以下のプライマー(5'-ATGCACTGTAAACTTTCGATCCCGTCTTAG-3'(配列番号211)、5'-CTACAGGGTGTGACGCTATGGTCAG-3'(配列番号212))を用いて形質転換体のゲノムDNAに対してPCRを行い、約4,700bpの増幅断片のみがみられたものをmpkBΔ候補株とし、最終的に定量RT-PCRによりmpkB遺伝子が発現していないことを確認した(図10)。
【0127】
(3)mpkBΔ株におけるマイクロアレイ解析
アスペルギルス・ニドランスABPU1 ΔligD::ptrA株及びmpkB破壊株の分生子を2x106個分生子/mlになるように、終濃度0.02μg/mlのビオチン、5mMのウリジン、10mMのウラシル、0.5μg/mlのピリドキシンを添加した200mlのCD液体培地を入れた500ml容の三角フラスコに接種し、37℃、24時間振盪培養した。菌体をMIRACLOTH(CALBIOCHEM社製)で集菌し、この回収した菌を乳鉢中で液体窒素を注ぎながらパウダー状になるまで粉砕した。10 mlのSepasol-RNA I Super(ナカライテスク社製)を入れた50 ml容チューブにパウダー状の菌体を移し懸濁後、室温にて5分間放置した。9,500×g、4℃、10分間遠心し、水層を15ml容チューブに移し、2 mlのクロロホルムを加え激しく撹拌後、室温にて5分間放置した。11,000×g、4℃、15分間遠心し、水層を別の15ml容チューブに移し、等量の水飽和酸性フェノール:クロロホルム(5:1)を加え激しく撹拌し、室温にて5分間放置した。10,000×g、4℃、10分間遠心し、水層を別の15ml容チューブに移し、半量のイソプロパノール、半量のHigh Salt Precipitation Solution(0.8M Sodium Citrate、1.2M NaCl、DEPC処理済み)を加え、撹拌後、室温にて10分間放置した。10,000×g、4℃、10分間遠心し、上清を捨て、70%エタノールでリンスした。風乾後、適量のDEPC(diethylpyrocarbonate)水に溶解し、total RNA溶液とした。次にoligotex-dt30 〈Super〉mRNA Purification kit (From Total RNA)(タカラバイオ社製)を用い、添付のマニュアルに従いtotal RNAからmRNAを精製した。
【0128】
マイクロアレイ解析をするにあたりプローブの作製は、Cy3、Cy5を用いたポストラベリング法でおこなった。精製したmRNAを1μg/9μlに調製し、CyScribe cDNA Post Labelling Kit(GE healthcare)を用い、添付のマニュアルに従ってmRNAからアミノアリル標識cDNAを合成した。合成したアミノアリル標識cDNAはCyScribe GFX Purification Kit(GE healthcare)を用いて精製し、続いてCyScribe cDNA Post Labelling Kit(GE healthcare)によりCyDye標識した。この時、ABPU1 ΔligD::ptrA株由来のcDNAをCy3で、mpkB破壊株由来のcDNAをCy5で標識した組と、その逆(Dyeスワップ)で標識した組とを準備した。標識したプローブはCyScribe GFX Purification Kit(GE healthcare)により精製し、遠心エバポレーターで乾燥した。表17に記すハイブリダイゼーション溶液を調製し、この溶液60μLを用いて上記組み合わせになるように、Cy3標識したcDNAを溶解させた後、その溶解液を用いてCy5で標識したcDNAを溶解させるといった手順で混合しハイブリダイゼーション溶液とした。
【0129】
【表17】
【0130】
アスペルギルス・ニドランスのマイクロアレイは、TIGERのガラススライドマイクロアレイ(version2)を使用した。プレハイブリダイゼーションバッファー(5×SSC、0.1% SDDS、1%BSA)を調製し、このバッファー中にガラススライドを浸し、少なくとも1時間、42℃で保温した。ガラススライドをMilliQ水で満たした洗浄用容器中に移し、2分間室温にて振盪洗浄した。MilliQ水での洗浄はMilliQ水2Lを使い切るまで繰り返した。次に別の容器にイソプロパノールを満たし、これにガラススライドを移して、2分間室温にて振盪洗浄した。その後800rpm、2分間遠心しイソプロパノールを完全に除去した。
【0131】
プレハイブリダイゼーション後のアレイスライドにSpaced Cover Glass XL (タカラバイオ社製)を被せ、アレイスライドとカバーガラスの間に前述の標識したハイブリダイゼーション溶液を充填させた。これをSlideBooster(Advaritix)に設置し、42℃で17時間保持した。その後SlideBoosterからアレイスライドを取り出し、Wash buffer 1(2×SSC、0.1% SDS)を満たした洗浄用容器中でカバーガラスを剥がした。アレイスライドをWash buffer 1中で15分間室温にて振盪洗浄した。次にWash Buffer 2(1×SSC、0.1% SDS)を満たした洗浄用容器に移し、5分間室温にて振盪洗浄した。最後にWash Buffer 3(0.2×SSC)を満たした洗浄用容器に移し、5分間室温にて振盪洗浄した後、800rpm、2分間遠心し水分を完全に除去した。
【0132】
アレイスライドの画像化にはGenePix4000B(Axon)を使用した。画像化したスキャンデータをGenePix4000Bに標準添付されているGenePixProマイクロアレイ解析ソフトを用いて数値化した。アレイデータはGenomic Profiler(三井情報(株))を用いて標準化し、Excelによりt検定をおこなった。ここで得られたデータをt検定のp値により絞り込むことによって、十分信頼性のおけるデータのみ後の解析に使用した。得られたアレイデータから、アスペルギルス・ニドランスABPU1 ΔligD::ptrA株とmpkB破壊株の間の発現量比の差が大きい遺伝子を抽出し、MpkB依存的に発現変動する遺伝子とした。
【0133】
〔実施例11〕
mpkBΔ株におけるsodM遺伝子の定量RT-PCR
(1)RNAの抽出
アスペルギルス・ニドランスABPU1 ΔligD::ptrA株及びmpkB破壊株の分生子を2x106個分生子/mlになるように、終濃度0.02μg/mlのビオチン、5mMのウリジン、10mMのウラシル、0.5μg/mlのピリドキシンを添加した100mlのCD液体培地を入れた300ml容の三角フラスコに接種し、37℃、24時間振盪培養した。菌体をMIRACLOTH(CALBIOCHEM社製)で集菌した。この回収した菌を乳鉢中で液体窒素を注ぎながらパウダー状になるまで粉砕した。10mlのSepasol-RNA I Super(ナカライテスク社製)を入れた50ml容チューブにパウダー状の菌体を移し懸濁後、室温にて5分間放置した。9,500×g、4℃、10分間遠心し、水層を15ml容チューブに移し、2mlのクロロホルムを加え激しく撹拌後、室温にて5分間放置した。11,000×g、4℃、15分間遠心し、水層を別の15ml容チューブに移し、等量の水飽和酸性フェノール:クロロホルム(5:1)を加え激しく撹拌し、室温にて5分間放置した。10,000×g、4℃、10分間遠心し、水層を別の15ml容チューブに移し、半量のイソプロパノール、半量のHigh Salt Precipitation Solution(0.8M Sodium Citrate、1.2M NaCl、DEPC処理済み)を加え、撹拌後、室温にて10分間放置した。10,000×g、4℃、10分間遠心し、上清を捨て、70%エタノールでリンスした。風乾後、適量のDEPC(diethylpyrocarbonate)水に溶解し、total RNA溶液とした。
【0134】
(2)cDNAの合成
cDNAの合成には、total RNAを用いた。260nmにおける吸光度からの計算値で100μgのtotal RNAをRNase freeの1.5mlチューブに移し、さらにDEPC処理水を加え87.5μlとした。これにRNase-free DNaseI set(QIAGEN)に付属のBuffer RDDを10μlとDNaseI solutionを10μl加え、37℃で10分間反応させて混荏するゲノムDNAの分解を行った。その後、RNeasy mini kit(QIAGEN)のRNA cleanupプロトコールに従ってtotal RNAをクリーンアップした。クリーンアップしたtotal RNA 2μg量をRNase freeの1.5mlチューブに移し、DEPC処理水を加え10μlとした。これにHigh Capacity cDNA Reverse Transcription kit(Applied Biosystems)に付属の反応液を表18に示す容量加え、20μlのcDNA合成反応液を調製した。
【0135】
【表18】
【0136】
調製したcDNA合成反応液を25℃で2分間保温し、プライマーをアニーリングさせた。その後、37℃で120分間保温し、逆転写反応させた。次に85℃で5秒間保温することにより、Multiscribe Reverse Transcriptaseを失活させた。反応終了後のcDNA溶液を1/10希釈し、次の定量RT-PCR実験に供した。
【0137】
(3)定量RT-PCR
合成したcDNAの蛍光色素によるラベリング反応には、DyNAmo SYBR Green qPCR Kit(FinnzymeS社製)を用いた。スキャナー解析及びデータ解析には、MiniOpticonリアルタイムPCR解析システム(バイオ・ラッドラボラトリーズ(株))を用いた。ラベリング反応操作は、DyNAmo SYBR Green qPCR Kit添付マニュアルにしたがった。通常の生育条件でのmpkBΔ株とコントロール株におけるsodM遺伝子の転写量の変化を調べた。コントロールをHistone H2Bとし、それぞれ以下の特異的プライマーを用いた。
Histone H2B FW:5'-CACCCGGACACTGGTATCTC-3'(配列番号213)
Histone H2B RW:5'-GAATACTTCGTAACGGCCTTGG-3'(配列番号214)
sodM FW:5'-CTCTGTAGAGGCGTTCATCAAG-3'(配列番号215)
sodM RW:5'-ACTGCAAGTAATACGCATGCTC-3'(配列番号216)
【0138】
1.5mlチューブに、5μlのcDNA(cDNA合成時に用いたtotalRNA量で100ng相当)、300nMの各標的遺伝子特異プライマー、10μlの2xMasterMix、滅菌水を加え25μlとした。専用のPCRチューブ8-Strip Low Profile Tubesに分注し、8-Strip Ultra Clear Caps(共にMJResearch社)にてふたをし、MiniOpticonリアルタイムPCR解析システムにセットした。反応条件は95℃、10分間のヒートショック後、熱変性95℃、10秒、アニーリング60℃、30秒、伸長反応72℃、30秒、プレートリードの反応を40サイクル行い、60〜95℃のグラジェント(0.2℃おきに1秒間hold)でMelting Curveの作成を行った。設定はHistone H2Bをスタンダード、sodMをサンプルとして設定した。PCRは各cDNA、各標的遺伝子につき三連で行った。PCRを行うにあたり、各標的遺伝子特異プライマー対と2×Master Mixのみの反応系を組み、95℃、10分間のヒートショック後、熱変性95℃、10秒、60〜95℃のグラジェントで50秒、プレートリードの反応を40サイクル行い、50〜95℃のグラジェントで(0.5℃おきに2秒間hold)でMelting Curveの作成を行い、プライマー同士の非特異的な増幅が起きないことを確認した。PCR終了後、MiniOpticonリアルタイムPCR解析システム付属の専用解析ソフトを用いて解析を行った。Quantitation画面上で各遺伝子のPCR産物が対数的に増幅し始めるサイクル数(CT)を決定した。この値をもとに各cDNAの標的遺伝子の相対発現量を以下の計算方法から求めた。
a)△CT値の算出(各cDNAの標的遺伝子とHistone H2Bとの差)
△CT=(y−x)
xはCT(Histone H2B)であり、yはCT(Target gene)である。
b)△CT値の算出(あるcDNAの△CTと基準とするcDNAの△CTとの差)
△CT=△CT(C)−△CT(S)
CはComparative cDNAであり、SはStandard cDNAである。
C)相対発現量の算出
2-(△CT)
また、Melting Curveの波形から正しく増幅されていることを確認し、さらにPCR産物をアガロースゲル電気泳動に供し、目的の大きさに増幅されていることを確認した。
【0139】
RT-PCRの結果を図10に、Histone H2Bとの発現量比としてグラフ化したものを示した。mpkBΔ株におけるsodM遺伝子の発現量はコントロール株と比べて優位に増加した。すなわち、sodM遺伝子の転写量の変化はMpkBに依存していると考えられた。よって、sodM遺伝子の5'-UTR(5'上流領域のプロモーター)はレポーター遺伝子と機能的に結合させることによって、本発明のスクリーニング方法において、被検物質のMpkB経路の阻害作用を検出することができる。
【0140】
〔実施例12〕
アスペルギルス・オリゼ(Aspergillus oryzae)のMpkA依存的遺伝子の決定
(1)RNA抽出
アスペルギルス・オリゼのmpkA遺伝子破壊株(NS4 ΔligD::ptrA ΔmpkA::sC)及びコントロール株(NS4 ΔligD::ptrA sC+)は酒類総合研究所の水谷治博士より供与いただいた(Mizutani et al. Fungal Genet. Biol., 2008 (45) 878-889)。mpkA遺伝子破壊株及びコントロール株の分生子を2×106個分生子/mlになるように、窒素源をグルタミン酸ナトリウムに置き換えた200 mlのCD液体培地を入れた500 ml容の三角フラスコに接種し、30℃、24時間振盪培養した。
【0141】
菌体をMIRACLOTH(CALBIOCHEM社製)で集菌し、この回収した菌を乳鉢中で液体窒素を注ぎながらパウダー状になるまで粉砕した。10mlのSepasol-RNA I Super(ナカライテスク)を入れた50ml容チューブにパウダー状の菌体を移し懸濁後、室温にて5分間放置した。9,500×g、4℃、10分間遠心し、水層を15ml容チューブに移し、2mlのクロロホルムを加え激しく撹拌後、室温にて5分間放置した。11,000×g、4℃、15分間遠心し、水層を別の15ml容チューブに移し、等量の水飽和酸性フェノール:クロロホルム(5:1)を加え激しく撹拌し、室温にて5分間放置した。10,000×g、4℃、10分間遠心し、水層を別の15ml容チューブに移し、半量のイソプロパノール、半量のHigh Salt Precipitation Solution(0.8M Sodium Citrate、1.2M NaCl、DEPC処理済み)を加え、撹拌後、室温にて10分間放置した。10,000×g、4℃、10分間遠心し、上清を捨て、70%エタノールでリンスした。風乾後、適量のDEPC(diethylpyrocarbonate)水に溶解し、total RNA溶液とした。次にoligotex-dt30 〈Super〉mRNA Purification kit (From Total RNA)(タカラバイオ社製)を用い、添付のマニュアルに従いtotal RNAからmRNAを精製した。
【0142】
マイクロアレイ解析をするにあたりプローブの作製は、Cy3、Cy5を用いたポストラベリング法でおこなった。精製したmRNAを1μg/9μlに調製し、CyScribe cDNA Post Labelling Kit(GE healthcare社製)を用い、添付のマニュアルに従ってmRNAからアミノアリル標識cDNAを合成した。合成したアミノアリル標識cDNAはCyScribe GFX Purification Kit(GE healthcare社製)を用いて精製し、続いてCyScribe cDNA Post Labelling Kit(GE healthcare社製)によりCyDye標識した。この時、コントロール株由来のcDNAをCy3で、mpkA破壊株由来のcDNAをCy5で標識した組と、その逆(Dyeスワップ)で標識した組とを準備した。標識したプローブはCyScribe GFX Purification Kit(GE healthcare社製)により精製し、遠心エバポレーターで乾燥した。実施例10の表17に示したハイブリダイゼーション溶液を調製し、この溶液60μLを用いて上記組み合わせになるように、Cy3標識したcDNAを溶解させた後、その溶解液を用いてCy5で標識したcDNAを溶解させるといった手順で混合しハイブリダイゼーション溶液とした。
【0143】
アスペルギルス・オリゼのマイクロアレイは、ファームラボ社製の麹菌12Kガラススライドマイクロアレイを使用した。アレイスライドにギャップカバーガラス(マツナミ社製)を被せ、アレイスライドとカバーガラスの間に前述の標識したハイブリダイゼーション溶液を充填させた。アレイスライドチャンバー(タカラバイオ社製)に設置し、40℃に設定したウォーターバス中で17時間保持した。その後アレイスライドチャンバーからアレイスライドを取り出し、Wash buffer 1(2 ×SSC、0.03% SDS)を満たしたプラスチック製の洗浄用容器中でカバーガラスを剥がした。アレイスライドをWash buffer 1中で15分間室温にて振盪洗浄した。次にWash Buffer 2(0.2× SSC)を満たした洗浄用容器に移し、5分間室温にて振盪洗浄した。最後にWash Buffer 3(0.05 × SSC)を満たした洗浄用容器に移し、5分間室温にて振盪洗浄した後、800rpm、2分間遠心し水分を完全に除去した。
【0144】
アレイスライドの画像化にはGenePix4000B(Axon)を使用した。画像化したスキャンデータをGenePix4000Bに標準添付されているGenePixProマイクロアレイ解析ソフトを用いて数値化した。アレイデータはGenomic Profiler(三井情報(株))を用いて標準化し、Excelによりt検定をおこなった。ここで得られたデータをt検定のp値により絞り込むことによって、十分信頼性のおけるデータのみ後の解析に使用した。得られたアレイデータから、アスペルギルス・オリゼmpkA破壊株とコントロール株の間の発現量比の差が大きい遺伝子を抽出し、MpkA依存的に発現変動する遺伝子とした。
【0145】
その結果、アスペルギルス・オリゼのMpkA依存的に発現変動する遺伝子のうち、経路遮断により活性上昇する遺伝子(AO090038000214、AO090003001496、 AO090009000634、AO090102000339)、及び経路遮断により活性が抑制される遺伝子(AO090003001367、AO090012000623、AO090012000169、AO090012000625、AO090012000622、AO090003001336、AO09001000644)が特定された。これら遺伝子のプロモータは、レポーター遺伝子と機能的に結合させることによって、MAPキナーゼ経路の一つであるMpkA経路に対する被検物質の活性化作用又は阻害作用を検出することができる。
【0146】
〔実施例13〕
本実施例では、Fujioka et al.(2007) Eukaryotic Cell 6: 1497-1510 の論文中に記載されている、cell wall integrity 経路を構成するMAP キナーゼMpkAの破壊株と野生株における転写解析のデータをもとにagsA遺伝子及びagsB遺伝子のプロモータ領域を使用したレポーター株を作製した。
【0147】
(1)レポーター遺伝子ベクターの構築
Broad Institute のアスペルギルス属ゲノムデータベース(http://www.broad.mit.edu/annotation/genome/aspergillus_group/MultiHome.html)からアスペルギルス・ニドランス(Aspergillus nidulans)のagsA遺伝子及びagsB遺伝子の5'上流領域1,000 bpを表19に示すプライマーセットを用いてPCRで増幅した。
【0148】
【表19】
【0149】
PCR産物をpGEM-T Easy Vector(Promega社製)に導入し、pGEM-PagsA及びpGEM-PagsBを得た。次いでpGEM-PagsA及びpGEM-PagsBをNotIで消化し、PagsA及びPagsB(PagsA及びPagsBの5'上流領域1,000bP)を切り出し、pNA(N)EGFPのNotIサイトへ導入してpNA(N)EGFP/PagsA及びpNA(N)EGFP/PagsBを作製した。なおpNA(N)EGFPは東北大学大学院農学研究科・古川健太郎博士より供与して頂いたもので、EGFPに核移行型シグナルLacIが付加してある(Furukawa et al. Biosci. Biotechnol. Biochem., 2007 (71) 1724-1730)。
【0150】
上記プラスミドにおいて、pNA(N)EGFP/PagsAについてはオーレオバシジン耐性遺伝子aurA内に存在するSrfIサイトで、pNA(N)EGFP/PagsBについてはaurA内に存在するBssHIIサイトで直鎖状に切断した断片を用いて、A.nidulans A89+argB(正式にはRB89)株及びmpkAΔ株(A89 ΔmpkA::argB)に対して形質転換を行った。オーレオバシジン耐性株を選抜し、PCRにより遺伝子導入を確認した。
【0151】
(2)EGFP-LacI蛍光観察
96ウェルのタイタープレートにCDビオチン液体培地200μL、RB89株からの形質転換体から調製した胞子懸濁液20(200コ)の混合液を加え、30℃で24時間培養した。タイタープレートから菌体をこすり取りEGFP-LacIの蛍光を観察した。PagsB-EGFP形質転換体においては、核に局在したEGFP-LacI蛍光が観察されたが、PagsA-EGFP形質転換体では蛍光は観察されなかった。一方、mpkAΔ株を宿主とした場合には、PagsA-EGFP形質転換体では蛍光が観察され、PagsB-EGFP形質転換体では蛍光は観察されなかった(図11)。これはagsA、agsBの発現解析パターンと完全に一致するものである(Fujioka et al. Eukaryotic Cell, 2007 (6) 1497-1510)。以上の結果から、PagsA-EGFP及びPagsB-EGFPレポ一夕ーはMpkA経路のシグナル切断作動性薬剤のスクリーニングに利用できると言える。
【0152】
〔実施例14〕
cat1遺伝子のHOG経路レポーター遺伝子としての可能性検証
(1)ゲノムデータベースからのいもち病菌CAT-1遺伝子ホモログの探索
本実施例では、アカパンカビ(Neurospora crassa)においてCAT-1(catalase 1)遺伝子がHOG経路依存的に転写制御を受けるという報告に基づき、いもち病菌のゲノムデータベース (http://www.broad.mit.edu/annotation/genome/magnaporthe_grisea/MultiHome.html) からCAT-1遺伝子のオルソログを検索した。N. crassaのCAT-1遺伝子(NCU08791.3)をクエリー配列としてblastp検索を行ったところ、MGG_10061.6(Genbankアクセッション番号XM_365841)が高いホモロジーでヒットしたことからこの遺伝子をいもち病菌におけるcat1遺伝子とした。
【0153】
(2)total RNAの調製及びcDNAの合成
いもち病菌におけるcat1遺伝子がHOG経路構成的活性化剤フルジオキソニル又はイプロジオンを処理したときに、特異的に発現変化があり検出マーカーとして利用可能か否かを検証した。
【0154】
ブレンダーで細かく裁断したいもち病菌の菌体を50ml YG液体培地(0.4% yeast extract、2% glucose/200ml三角フラスコ)で27℃、120rpm、22h培養後、20mg/mlのフルジオキソニル(in DMSO)を50μl添加した(終濃度20μg/ml)。コントロールはDMSOを50μl添加した。薬剤添加後15分間培養を続け、ミラクロスで菌体を回収した。液体窒素を用いて菌体を破砕し5mlのTRIzol (Invitrogen社製)に懸濁後5分間静置した。1mlのクロロホルムを加え激しく攪拌後、室温で3分間静置した。5,000 rpm、15min、4℃で遠心し、RNAを含む約3mlの水層を別のチューブに移した。このRNA 画分のうち1.2mlをPureLink Micro-to-Midi Total RNA Purification Kit (Invitrogen社製)を用いて精製した。このtotal RNAをPrimeScript RT reagent Kit (Perfect Real Time)(タカラバイオ社製)を用いて逆転写し、一本鎖cDNAを作製した。逆転写のprimerにはOligo dT PrimerとRandom 6 mersの両方を用いた。
【0155】
(3)リアルタイムPCRによるcat1遺伝子の転写応答解析
内部標準の遺伝子にはβ-tubulin遺伝子(MGG_00604.6)を用いた。解析に用いたプライマーを以下に示す。
いもち病菌におけるcat1遺伝子用
Mgcat1 FW1: 5'-TGGATCAGTTCTTCCACAGCA-3'(配列番号221)
Mgcat1 RV1: 5'-AACTCCACCCGCTTTCTCC-3'(配列番号222)
β-tubulin遺伝子用
Mg_b-tubulin FW1: 5'-GAGTCCAACATGAACGATCT-3'(配列番号223)
Mg_b-tubulin RV1: 5'-GTACTCCTCTTCCTCCTCGT-3'(配列番号224)
【0156】
リアルタイムPCRの反応系の調製にはSYBR Premix Ex Taq II (Perfect Real Time) (タカラバイオ社製)を用い、thermal cyclerには Thermal Cycler Dice Real Time System (タカラバイオ社製)を使用した。反応条件は、初期変性95℃、30秒、その後、変性95℃、5秒、アニーリング/伸長:60℃、30秒(40サイクル)で行った。
【0157】
RT-PCRの結果を図12に示した。cat1遺伝子の転写量はフルジオキソニル及びイプロジオン処理により有意に増加していた。よって、いもち病菌におけるcat1遺伝子の5'-UTR(5'上流領域のプロモーター)はレポーター遺伝子と機能的に結合させることにより、HOG経路活性化剤のスクリーニング系として利用できる可能性が確認された。
【産業上の利用可能性】
【0158】
本発明は、感染性微生物に使用される抗菌剤を開発する分野に利用することができる。
【特許請求の範囲】
【請求項1】
被検物質を作用させた細胞における、表1に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる少なくとも1種の遺伝子の発現を測定する工程と、
【表1】
上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を呼吸鎖阻害剤の候補物質として同定する工程とを含む、スクリーニング方法。
【請求項2】
上記呼吸鎖阻害剤は、Complex1阻害剤、Complex2阻害剤、Complex3阻害剤、Complex5 (F1Fo ATPase)阻害剤、及び電子伝達に作用してATP合成を阻害する脱共役作用剤からなる群から選ばれる少なくとも1種の阻害剤と類似の作用機序を有する阻害剤であることを特徴とする請求項1記載のスクリーニング方法。
【請求項3】
いもち病菌に対する呼吸鎖阻害作用を示す呼吸鎖阻害剤の候補物質として同定することを特徴とする請求項1記載のスクリーニング方法。
【請求項4】
表1に示す遺伝子群のうち少なくともXM_001403469(MGG_12936)遺伝子の発現を測定することを特徴とする請求項1記載のスクリーニング方法。
【請求項5】
表2に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる遺伝子のプロモータ領域と、当該プロモータ領域の制御下に発現可能なレポーター遺伝子とを連結した組換えDNA。
【表2】
【請求項6】
上記プロモータ領域は、上記遺伝子における開始コドンの5'末端から上流2kbの領域であることを特徴とする請求項5記載の組換えDNA。
【請求項7】
請求項5又は6記載の組換えDNAを導入した形質転換細胞。
【請求項8】
請求項7記載の形質転換細胞と、当該形質転換細胞におけるレポーター遺伝子の産物を測定する測定試薬とを含む、呼吸鎖阻害剤のスクリーニングキット。
【請求項9】
被検物質を作用させた細胞における、表3に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる少なくとも1種の遺伝子の発現を測定する工程と、
【表3】
上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質をエルゴステロール生合成阻害剤の候補物質として同定する工程とを含む、スクリーニング方法。
【請求項10】
上記エルゴステロール生合成阻害剤は、オキスポコナゾール・フマル酸塩と類似の作用機序を有する阻害剤であることを特徴とする請求項9記載のスクリーニング方法。
【請求項11】
いもち病菌に対するエルゴステロール生合成阻害作用を示すエルゴステロール生合成阻害剤の候補物質として同定することを特徴とする請求項9記載のスクリーニング方法。
【請求項12】
表3に示す遺伝子群のうち少なくともXM_362183 (MGG_04628)遺伝子の発現を測定することを特徴とする請求項9記載のスクリーニング方法。
【請求項13】
表4に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる遺伝子のプロモータ領域と、当該プロモータ領域の制御下に発現可能なレポーター遺伝子とを連結した組換えDNA。
【表4】
【請求項14】
上記プロモータ領域は、上記遺伝子における開始コドンの5'末端から上流2kbの領域であることを特徴とする請求項13記載の組換えDNA。
【請求項15】
請求項13又は14記載の組換えDNAを導入した形質転換細胞。
【請求項16】
請求項15記載の形質転換細胞と、当該形質転換細胞におけるレポーター遺伝子の産物を測定する測定試薬とを含む、エルゴステロール生合成阻害剤のスクリーニングキット。
【請求項17】
被検物質を作用させた細胞における、表5に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる少なくとも1種の遺伝子の発現を測定する工程と、
【表5】
上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を細胞壁生合成阻害剤の候補物質として同定する工程とを含む、スクリーニング方法。
【請求項18】
上記細胞壁生合成阻害剤は、ミカファンギン及び/又はカルコフロール・ホワイトと類似の作用機序を有する阻害剤であることを特徴とする請求項17記載のスクリーニング方法。
【請求項19】
いもち病菌に対する細胞壁生合成阻害作用を示す細胞壁生合成阻害剤の候補物質として同定することを特徴とする請求項17記載のスクリーニング方法。
【請求項20】
表5に示す遺伝子群のうち少なくともXM_364794 (MGG_09639)及びXM_368552 (MGG_00692)遺伝子の発現を測定することを特徴とする請求項17記載のスクリーニング方法。
【請求項21】
表6に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる遺伝子のプロモータ領域と、当該プロモータ領域の制御下に発現可能なレポーター遺伝子とを連結した組換えDNA。
【表6】
【請求項22】
上記プロモータ領域は、上記遺伝子における開始コドンの5'末端から上流2kbの領域であることを特徴とする請求項21記載の組換えDNA。
【請求項23】
請求項21又は22記載の組換えDNAを導入した形質転換細胞。
【請求項24】
請求項23記載の形質転換細胞と、当該形質転換細胞におけるレポーター遺伝子の産物を測定する測定試薬とを含む、細胞壁生合成阻害剤のスクリーニングキット。
【請求項25】
被検物質を作用させた細胞における、スーパーオキシドデスムターゼ遺伝子(Genbankアクセッション番号:XM_653297、配列番号71)の発現を測定する工程と、
上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質をMpkB経路阻害剤の候補物質として同定する工程とを含む、スクリーニング方法。
【請求項26】
上記MpkB経路阻害剤は、mpkB遺伝子又は当該遺伝子産物の阻害剤と類似の作用機序を有する阻害剤であることを特徴とする請求項25記載のスクリーニング方法。
【請求項27】
麹菌に対するMpkB経路阻害作用を示すMpkB経路阻害剤の候補物質として同定することを特徴とする請求項25記載のスクリーニング方法。
【請求項28】
スーパーオキシドデスムターゼ遺伝子(Genbankアクセッション番号:XM_653297、配列番号71)のプロモータ領域と、当該プロモータ領域の制御下に発現可能なレポーター遺伝子とを連結した組換えDNA。
【請求項29】
上記プロモータ領域は、上記遺伝子における開始コドンの5'末端から上流2kbの領域であることを特徴とする請求項28記載の組換えDNA。
【請求項30】
請求項28又は29記載の組換えDNAを導入した形質転換細胞。
【請求項31】
請求項30記載の形質転換細胞と、当該形質転換細胞におけるレポーター遺伝子の産物を測定する測定試薬とを含む、MpkB経路阻害剤のスクリーニングキット。
【請求項32】
被検物質を作用させた細胞における、表4に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる少なくとも1種の遺伝子の発現を測定する工程と、
【表7】
上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を細胞壁構築(cell wall integrity)経路阻害剤の候補物質として同定する工程とを含む、スクリーニング方法。
【請求項33】
上記細胞壁構築(cell wall integrity)経路阻害剤は、mpkA遺伝子又は当該遺伝子産物の阻害剤と類似の作用機序を有する阻害剤であることを特徴とする請求項32記載のスクリーニング方法。
【請求項34】
麹菌(Aspergillus oryzae)に対する細胞壁構築(cell wall integrity)経路阻害作用を示す細胞壁構築(cell wall integrity)経路阻害剤の候補物質として同定することを特徴とする請求項32記載のスクリーニング方法。
【請求項35】
表7に示す遺伝子群のうち少なくともXM_001825115 (AO090038000214)遺伝子の発現を測定することを特徴とする請求項32記載のスクリーニング方法。
【請求項36】
表8に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる遺伝子のプロモータ領域と、当該プロモータ領域の制御下に発現可能なレポーター遺伝子とを連結した組換えDNA。
【表8】
【請求項37】
上記プロモータ領域は、上記遺伝子における開始コドンの5'末端から上流2kbの領域であることを特徴とする請求項36記載の組換えDNA。
【請求項38】
請求項36又は37記載の組換えDNAを導入した形質転換細胞。
【請求項39】
請求項38記載の形質転換細胞と、当該形質転換細胞におけるレポーター遺伝子の産物を測定する測定試薬とを含む、細胞壁構築(cell wall integrity)経路阻害剤のスクリーニングキット。
【請求項40】
被検物質を作用させた細胞における、agsA遺伝子(Genbankアクセッション番号:XM_658397、配列番号83)及び/又はagsB遺伝子(Genbankアクセッション番号:XM_655819、配列番号84)の発現を測定する工程と、
上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を細胞壁構築(cell wall integrity)経路阻害剤の候補物質として同定する工程とを含む、スクリーニング方法。
【請求項41】
上記細胞壁構築(cell wall integrity)経路阻害剤は、上記遺伝子又は当該遺伝子産物の阻害剤と類似の作用機序を有する阻害剤であることを特徴とする請求項40記載のスクリーニング方法。
【請求項42】
Aspergillus nidulansに対する細胞壁構築(cell wall integrity)経路阻害作用を示す細胞壁構築(cell wall integrity)経路阻害剤の候補物質として同定することを特徴とする請求項40記載のスクリーニング方法。
【請求項43】
agsA遺伝子及びagsB遺伝子のうち少なくともagsB遺伝子の発現を測定することを特徴とする請求項40記載のスクリーニング方法。
【請求項44】
agsA遺伝子(Genbankアクセッション番号:XM_658397、配列番号83)及び/又はagsB遺伝子(Genbankアクセッション番号:XM_655819、配列番号84)のプロモータ領域と、当該プロモータ領域の制御下に発現可能なレポーター遺伝子とを連結した組換えDNA。
【請求項45】
上記プロモータ領域は、上記遺伝子における開始コドンの5'末端から上流2kbの領域であることを特徴とする請求項44記載の組換えDNA。
【請求項46】
請求項44又は45記載の組換えDNAを導入した形質転換細胞。
【請求項47】
請求項46記載の形質転換細胞と、当該形質転換細胞におけるレポーター遺伝子の産物を測定する測定試薬とを含む、細胞壁構築(cell wall integrity)経路阻害剤のスクリーニングキット。
【請求項48】
被検物質を作用させた細胞における、cat1遺伝子(Genbankアクセッション番号:XM_365841、配列番号85)の発現を測定する工程と、
上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を浸透圧応答(HOG: high osmolarity glycerol)経路活性化剤の候補物質として同定する工程とを含む、スクリーニング方法。
【請求項49】
上記浸透圧応答(HOG: high osmolarity glycerol)経路活性化剤は、フルジオキソニル及び/又はイプロジオンと類似の作用機序を有する活性化剤であることを特徴とする請求項48記載のスクリーニング方法。
【請求項50】
いもち病菌に対する浸透圧応答(HOG: high osmolarity glycerol)経路活性作用を示す浸透圧応答(HOG: high osmolarity glycerol)経路活性化剤の候補物質として同定することを特徴とする請求項48記載のスクリーニング方法。
【請求項51】
cat1遺伝子(Genbankアクセッション番号:XM_365841、配列番号85)のプロモータ領域と、当該プロモータ領域の制御下に発現可能なレポーター遺伝子とを連結した組換えDNA。
【請求項52】
上記プロモータ領域は、上記遺伝子における開始コドンの5'末端から上流1.5kbの領域であることを特徴とする請求項51記載の組換えDNA。
【請求項53】
請求項51又は52記載の組換えDNAを導入した形質転換細胞。
【請求項54】
請求項53記載の形質転換細胞と、当該形質転換細胞におけるレポーター遺伝子の産物を測定する測定試薬とを含む、浸透圧応答(HOG: high osmolarity glycerol)経路活性化剤のスクリーニングキット。
【請求項1】
被検物質を作用させた細胞における、表1に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる少なくとも1種の遺伝子の発現を測定する工程と、
【表1】
上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を呼吸鎖阻害剤の候補物質として同定する工程とを含む、スクリーニング方法。
【請求項2】
上記呼吸鎖阻害剤は、Complex1阻害剤、Complex2阻害剤、Complex3阻害剤、Complex5 (F1Fo ATPase)阻害剤、及び電子伝達に作用してATP合成を阻害する脱共役作用剤からなる群から選ばれる少なくとも1種の阻害剤と類似の作用機序を有する阻害剤であることを特徴とする請求項1記載のスクリーニング方法。
【請求項3】
いもち病菌に対する呼吸鎖阻害作用を示す呼吸鎖阻害剤の候補物質として同定することを特徴とする請求項1記載のスクリーニング方法。
【請求項4】
表1に示す遺伝子群のうち少なくともXM_001403469(MGG_12936)遺伝子の発現を測定することを特徴とする請求項1記載のスクリーニング方法。
【請求項5】
表2に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる遺伝子のプロモータ領域と、当該プロモータ領域の制御下に発現可能なレポーター遺伝子とを連結した組換えDNA。
【表2】
【請求項6】
上記プロモータ領域は、上記遺伝子における開始コドンの5'末端から上流2kbの領域であることを特徴とする請求項5記載の組換えDNA。
【請求項7】
請求項5又は6記載の組換えDNAを導入した形質転換細胞。
【請求項8】
請求項7記載の形質転換細胞と、当該形質転換細胞におけるレポーター遺伝子の産物を測定する測定試薬とを含む、呼吸鎖阻害剤のスクリーニングキット。
【請求項9】
被検物質を作用させた細胞における、表3に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる少なくとも1種の遺伝子の発現を測定する工程と、
【表3】
上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質をエルゴステロール生合成阻害剤の候補物質として同定する工程とを含む、スクリーニング方法。
【請求項10】
上記エルゴステロール生合成阻害剤は、オキスポコナゾール・フマル酸塩と類似の作用機序を有する阻害剤であることを特徴とする請求項9記載のスクリーニング方法。
【請求項11】
いもち病菌に対するエルゴステロール生合成阻害作用を示すエルゴステロール生合成阻害剤の候補物質として同定することを特徴とする請求項9記載のスクリーニング方法。
【請求項12】
表3に示す遺伝子群のうち少なくともXM_362183 (MGG_04628)遺伝子の発現を測定することを特徴とする請求項9記載のスクリーニング方法。
【請求項13】
表4に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる遺伝子のプロモータ領域と、当該プロモータ領域の制御下に発現可能なレポーター遺伝子とを連結した組換えDNA。
【表4】
【請求項14】
上記プロモータ領域は、上記遺伝子における開始コドンの5'末端から上流2kbの領域であることを特徴とする請求項13記載の組換えDNA。
【請求項15】
請求項13又は14記載の組換えDNAを導入した形質転換細胞。
【請求項16】
請求項15記載の形質転換細胞と、当該形質転換細胞におけるレポーター遺伝子の産物を測定する測定試薬とを含む、エルゴステロール生合成阻害剤のスクリーニングキット。
【請求項17】
被検物質を作用させた細胞における、表5に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる少なくとも1種の遺伝子の発現を測定する工程と、
【表5】
上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を細胞壁生合成阻害剤の候補物質として同定する工程とを含む、スクリーニング方法。
【請求項18】
上記細胞壁生合成阻害剤は、ミカファンギン及び/又はカルコフロール・ホワイトと類似の作用機序を有する阻害剤であることを特徴とする請求項17記載のスクリーニング方法。
【請求項19】
いもち病菌に対する細胞壁生合成阻害作用を示す細胞壁生合成阻害剤の候補物質として同定することを特徴とする請求項17記載のスクリーニング方法。
【請求項20】
表5に示す遺伝子群のうち少なくともXM_364794 (MGG_09639)及びXM_368552 (MGG_00692)遺伝子の発現を測定することを特徴とする請求項17記載のスクリーニング方法。
【請求項21】
表6に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる遺伝子のプロモータ領域と、当該プロモータ領域の制御下に発現可能なレポーター遺伝子とを連結した組換えDNA。
【表6】
【請求項22】
上記プロモータ領域は、上記遺伝子における開始コドンの5'末端から上流2kbの領域であることを特徴とする請求項21記載の組換えDNA。
【請求項23】
請求項21又は22記載の組換えDNAを導入した形質転換細胞。
【請求項24】
請求項23記載の形質転換細胞と、当該形質転換細胞におけるレポーター遺伝子の産物を測定する測定試薬とを含む、細胞壁生合成阻害剤のスクリーニングキット。
【請求項25】
被検物質を作用させた細胞における、スーパーオキシドデスムターゼ遺伝子(Genbankアクセッション番号:XM_653297、配列番号71)の発現を測定する工程と、
上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質をMpkB経路阻害剤の候補物質として同定する工程とを含む、スクリーニング方法。
【請求項26】
上記MpkB経路阻害剤は、mpkB遺伝子又は当該遺伝子産物の阻害剤と類似の作用機序を有する阻害剤であることを特徴とする請求項25記載のスクリーニング方法。
【請求項27】
麹菌に対するMpkB経路阻害作用を示すMpkB経路阻害剤の候補物質として同定することを特徴とする請求項25記載のスクリーニング方法。
【請求項28】
スーパーオキシドデスムターゼ遺伝子(Genbankアクセッション番号:XM_653297、配列番号71)のプロモータ領域と、当該プロモータ領域の制御下に発現可能なレポーター遺伝子とを連結した組換えDNA。
【請求項29】
上記プロモータ領域は、上記遺伝子における開始コドンの5'末端から上流2kbの領域であることを特徴とする請求項28記載の組換えDNA。
【請求項30】
請求項28又は29記載の組換えDNAを導入した形質転換細胞。
【請求項31】
請求項30記載の形質転換細胞と、当該形質転換細胞におけるレポーター遺伝子の産物を測定する測定試薬とを含む、MpkB経路阻害剤のスクリーニングキット。
【請求項32】
被検物質を作用させた細胞における、表4に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる少なくとも1種の遺伝子の発現を測定する工程と、
【表7】
上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を細胞壁構築(cell wall integrity)経路阻害剤の候補物質として同定する工程とを含む、スクリーニング方法。
【請求項33】
上記細胞壁構築(cell wall integrity)経路阻害剤は、mpkA遺伝子又は当該遺伝子産物の阻害剤と類似の作用機序を有する阻害剤であることを特徴とする請求項32記載のスクリーニング方法。
【請求項34】
麹菌(Aspergillus oryzae)に対する細胞壁構築(cell wall integrity)経路阻害作用を示す細胞壁構築(cell wall integrity)経路阻害剤の候補物質として同定することを特徴とする請求項32記載のスクリーニング方法。
【請求項35】
表7に示す遺伝子群のうち少なくともXM_001825115 (AO090038000214)遺伝子の発現を測定することを特徴とする請求項32記載のスクリーニング方法。
【請求項36】
表8に示すGenbankアクセッション番号で特定される遺伝子群から選ばれる遺伝子のプロモータ領域と、当該プロモータ領域の制御下に発現可能なレポーター遺伝子とを連結した組換えDNA。
【表8】
【請求項37】
上記プロモータ領域は、上記遺伝子における開始コドンの5'末端から上流2kbの領域であることを特徴とする請求項36記載の組換えDNA。
【請求項38】
請求項36又は37記載の組換えDNAを導入した形質転換細胞。
【請求項39】
請求項38記載の形質転換細胞と、当該形質転換細胞におけるレポーター遺伝子の産物を測定する測定試薬とを含む、細胞壁構築(cell wall integrity)経路阻害剤のスクリーニングキット。
【請求項40】
被検物質を作用させた細胞における、agsA遺伝子(Genbankアクセッション番号:XM_658397、配列番号83)及び/又はagsB遺伝子(Genbankアクセッション番号:XM_655819、配列番号84)の発現を測定する工程と、
上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を細胞壁構築(cell wall integrity)経路阻害剤の候補物質として同定する工程とを含む、スクリーニング方法。
【請求項41】
上記細胞壁構築(cell wall integrity)経路阻害剤は、上記遺伝子又は当該遺伝子産物の阻害剤と類似の作用機序を有する阻害剤であることを特徴とする請求項40記載のスクリーニング方法。
【請求項42】
Aspergillus nidulansに対する細胞壁構築(cell wall integrity)経路阻害作用を示す細胞壁構築(cell wall integrity)経路阻害剤の候補物質として同定することを特徴とする請求項40記載のスクリーニング方法。
【請求項43】
agsA遺伝子及びagsB遺伝子のうち少なくともagsB遺伝子の発現を測定することを特徴とする請求項40記載のスクリーニング方法。
【請求項44】
agsA遺伝子(Genbankアクセッション番号:XM_658397、配列番号83)及び/又はagsB遺伝子(Genbankアクセッション番号:XM_655819、配列番号84)のプロモータ領域と、当該プロモータ領域の制御下に発現可能なレポーター遺伝子とを連結した組換えDNA。
【請求項45】
上記プロモータ領域は、上記遺伝子における開始コドンの5'末端から上流2kbの領域であることを特徴とする請求項44記載の組換えDNA。
【請求項46】
請求項44又は45記載の組換えDNAを導入した形質転換細胞。
【請求項47】
請求項46記載の形質転換細胞と、当該形質転換細胞におけるレポーター遺伝子の産物を測定する測定試薬とを含む、細胞壁構築(cell wall integrity)経路阻害剤のスクリーニングキット。
【請求項48】
被検物質を作用させた細胞における、cat1遺伝子(Genbankアクセッション番号:XM_365841、配列番号85)の発現を測定する工程と、
上記遺伝子の発現が、上記被検物質を作用させない場合の発現と比較して変動した場合に当該被検物質を浸透圧応答(HOG: high osmolarity glycerol)経路活性化剤の候補物質として同定する工程とを含む、スクリーニング方法。
【請求項49】
上記浸透圧応答(HOG: high osmolarity glycerol)経路活性化剤は、フルジオキソニル及び/又はイプロジオンと類似の作用機序を有する活性化剤であることを特徴とする請求項48記載のスクリーニング方法。
【請求項50】
いもち病菌に対する浸透圧応答(HOG: high osmolarity glycerol)経路活性作用を示す浸透圧応答(HOG: high osmolarity glycerol)経路活性化剤の候補物質として同定することを特徴とする請求項48記載のスクリーニング方法。
【請求項51】
cat1遺伝子(Genbankアクセッション番号:XM_365841、配列番号85)のプロモータ領域と、当該プロモータ領域の制御下に発現可能なレポーター遺伝子とを連結した組換えDNA。
【請求項52】
上記プロモータ領域は、上記遺伝子における開始コドンの5'末端から上流1.5kbの領域であることを特徴とする請求項51記載の組換えDNA。
【請求項53】
請求項51又は52記載の組換えDNAを導入した形質転換細胞。
【請求項54】
請求項53記載の形質転換細胞と、当該形質転換細胞におけるレポーター遺伝子の産物を測定する測定試薬とを含む、浸透圧応答(HOG: high osmolarity glycerol)経路活性化剤のスクリーニングキット。
【図1】

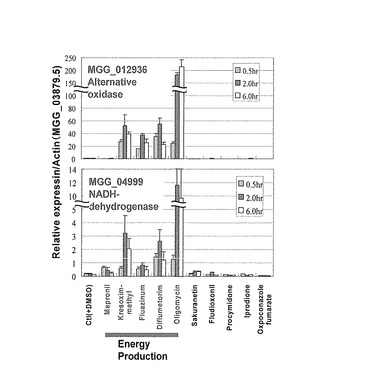
【図2】
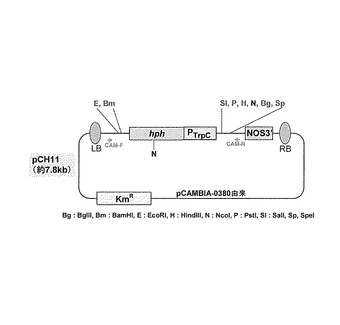
【図3】
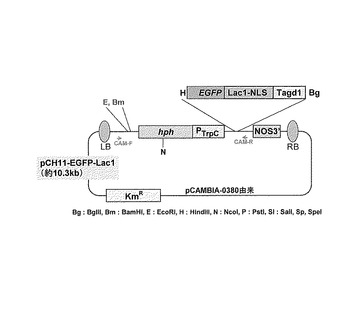
【図4】
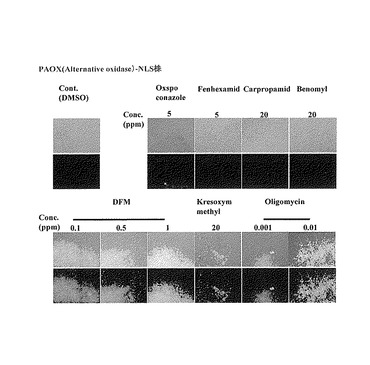
【図5】
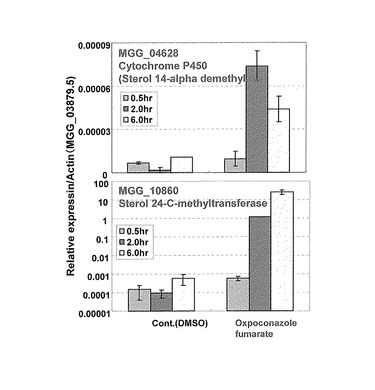
【図6】
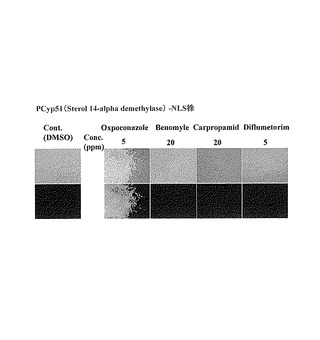
【図7】
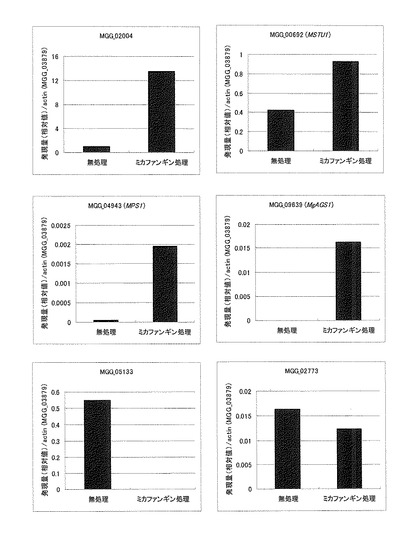
【図8】
【図9】

【図10】
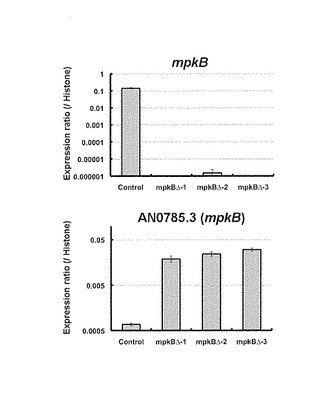
【図11】
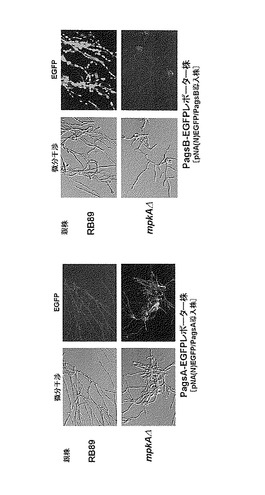
【図12】
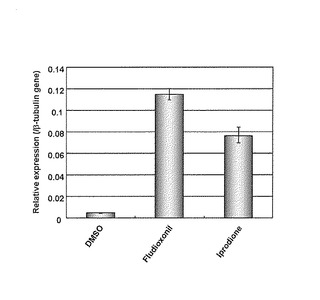
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公開番号】特開2010−220590(P2010−220590A)
【公開日】平成22年10月7日(2010.10.7)
【国際特許分類】
【出願番号】特願2009−74728(P2009−74728)
【出願日】平成21年3月25日(2009.3.25)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 刊行物名 :平成21年度 日本植物病理学会大会 プログラム・講演要旨予稿集 発行日 :平成21年3月9日 発行者 :日本植物病理学会 該当ページ:第68、及び74ページ 講演番号 :206、及び223
【出願人】(501167644)独立行政法人農業生物資源研究所 (200)
【出願人】(504157024)国立大学法人東北大学 (2,297)
【出願人】(000000169)クミアイ化学工業株式会社 (86)
【Fターム(参考)】
【公開日】平成22年10月7日(2010.10.7)
【国際特許分類】
【出願日】平成21年3月25日(2009.3.25)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 刊行物名 :平成21年度 日本植物病理学会大会 プログラム・講演要旨予稿集 発行日 :平成21年3月9日 発行者 :日本植物病理学会 該当ページ:第68、及び74ページ 講演番号 :206、及び223
【出願人】(501167644)独立行政法人農業生物資源研究所 (200)
【出願人】(504157024)国立大学法人東北大学 (2,297)
【出願人】(000000169)クミアイ化学工業株式会社 (86)
【Fターム(参考)】
[ Back to top ]