
無機ポリシラザン、これを含有してなるシリカ膜形成用塗布液及びシリカ膜の形成方法
【課題】水蒸気等の酸化剤中における焼成工程での収縮が小さく、シリカ膜の亀裂や半導体基板との剥離が発生しにくい無機ポリシラザン及び無機ポリシラザンを含有するシリカ膜形成用塗布液を提供すること。
【解決手段】1H―NMRスペクトルにおいて、4.75ppm以上で5.4ppm未満の範囲のピーク面積をAとし、4.5ppm以上で4.75ppm未満の範囲のピーク面積をBとし、4.2ppm以上で4.5ppm未満の範囲のピーク面積をCとしたとき、A/(B+C)の値が0.9〜1.5であり、(A+B)/Cの値が4.2〜50であり、ポリスチレン換算値による質量平均分子量が2000〜20000である無機ポリシラザンをシリカ膜形成用塗布液に含有させる。
【解決手段】1H―NMRスペクトルにおいて、4.75ppm以上で5.4ppm未満の範囲のピーク面積をAとし、4.5ppm以上で4.75ppm未満の範囲のピーク面積をBとし、4.2ppm以上で4.5ppm未満の範囲のピーク面積をCとしたとき、A/(B+C)の値が0.9〜1.5であり、(A+B)/Cの値が4.2〜50であり、ポリスチレン換算値による質量平均分子量が2000〜20000である無機ポリシラザンをシリカ膜形成用塗布液に含有させる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、特定の構成の無機ポリシラザン、当該無機ポリシラザンと有機溶剤とを必須成分として含有してなるシリカ膜形成用塗布液及びこれを用いたシリカ膜の形成方法に関する。
【背景技術】
【0002】
酸化珪素を主成分とするシリカ膜は、絶縁性、耐熱性、耐摩耗性及び耐蝕性の点で優れていることから、ハードコート材及び半導体装置の絶縁膜として広く使用されている。半導体装置の微細化に伴い、狭い隙間を埋め込む絶縁膜材料が望まれている。半導体装置に用いられる絶縁膜は、例えばCVD(Chemical Vapor Deposition、化学気相成長)法や塗布法により形成される。塗布法は、コストと生産性の面で優れていることから、品質の向上を目指し種々の材料が検討されている。
【0003】
ポリシラザンは、−SiH2−NH−を基本ユニットとする高分子化合物であり、比較的安価な方法である塗布法により、狭い隙間にも品質のよい酸化珪素を主成分とするシリカ膜を形成することができる。
【0004】
ポリシラザンを用いてシリカ膜を形成する方法としては、(1)ポリシラザンのキシレンやジブチルエーテル等の溶液を半導体基板等にスピンコート等により塗布する塗布工程、(2)ポリシラザンが塗布された半導体基板等を150℃程度に加熱し溶媒を蒸発させる乾燥工程、(3)この半導体基板等を水蒸気等の酸化剤の存在下において230〜900℃程度で焼成する焼成工程による方法(例えば、特許文献1及び2を参照)が知られている。ポリシラザンは、水蒸気における焼成工程でシリカに転化される。
尚、焼成工程において、ポリシラザンが、酸化剤である水蒸気によりシリカに転化される反応は、下記反応式(1)及び反応式(2)で表されることが知られている(例えば、非特許文献1参照)。
【化1】

【0005】
ポリシラザンを用いたシリカ膜の形成では、ポリシラザン塗膜がシリカ膜に変化する過程で収縮が起こる。ポリシラザンからシリカへの反応性を高めるとともに、シリカ表面のシラノール基(Si−OH)を減少させ絶縁性を向上させるためには、水蒸気中における焼成工程をより高温で行うことが好ましいが、高温における焼成は、このような収縮も大きくなる。水蒸気中における焼成工程での収縮率が高い場合には、シリカ膜の亀裂や、シリカ膜の半導体基板からの剥離が発生する場合があり、特に、半導体装置の素子間隔が狭い隙間を埋め込む素子間分離用途に無機ポリシラザンを使用し、高温で焼成する場合に、亀裂や剥離が発生しやすいという問題があった。今後、半導体素子の間隔を、更に狭めた半導体装置が要求されていることから、収縮の抑制された無機ポリシラザンが求められている。
【0006】
特許文献3には、一分子中のSiH3基に対するSiH2基の比が2.5〜8.4であり、元素比率でSi:N:H=50〜70質量%:20〜34質量%:5〜9質量%であるポリシラザンが耐熱性、耐摩耗性及び耐薬品性に優れると共に、表面硬度の高い被膜を与え、セラミックス成形体、特にセラミックス成形焼結体用のバインダーとして好適に使用できることが開示されている。しかしながら、このようなポリシラザンは、SiH3基の含量が多いために水蒸気中における焼成工程での収縮が大きく、500℃以上で焼成する場合にはシリカ膜の亀裂が発生しやすくなるという問題があった。
【0007】
特許文献4には、1H―NMRスペクトルのピーク面積比における、SiH1、SiH2及びSiH3の和に対するSiH3の割合が0.13〜0.45であり、数平均分子量200〜100,000であるポリシラザンを必須成分とする紫外線遮蔽ガラスの保護膜形成用組成物を、ガラス平面上の紫外線遮蔽層に塗布し、乾燥空気中で加熱することにより力学的強度や化学的安定に優れた保護膜が形成されることが開示されている。
【0008】
特許文献5には、1H―NMRスペクトルのピーク面積比における、SiH1とSiH2の和に対するSiH3の割合を0.15〜0.45に調整したポリシラザンの不活性有機溶剤溶液から成る層間絶縁膜形成用塗布液が、保存安定性及び塗布特性に優れると共に、絶縁性が高く、ち密で表面形状の良好な被膜を再現性よく形成することができることが開示されている。また、ポリシラザンの活性水素の一部をトリメチルシリル基に置換することにより調整が可能であり、調整剤としてヘキサメチルジシラザンを使用することが開示されている。しかしながら、ヘキサメチルジシラザンを反応させたポリシラザンは、水蒸気中における焼成工程での収縮が大きく、500℃以上で焼成する場合には、シリカ膜の亀裂が発生しやすくなるという問題があった。
【0009】
特許文献6には、1H−NMRスペクトルにおいて、SiH3基に由来する4.3〜4.5ppmのピーク面積に対する、SiH1基とSiH2基とに由来する4.5〜5.3ppmのピーク面積の比が、4.2〜50である無機ポリシラザンと、有機溶媒とを含有することを特徴とする絶縁膜形成用塗布液が、水蒸気中における焼成工程での収縮が小さく、シリカ被膜の亀裂や半導体基板との剥離が発生しにくい絶縁膜形成用塗布液、それを用いた絶縁膜及びそれに用いる化合物の製造方法を提供することが開示されている。しかしながら、シリカ膜中の残留カーボン低減のためには、高温焼成が求められる場合があり、更なる熱収縮改善の要求がある。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特開平7−223867号公報
【特許文献2】特開2001−308090号公報
【特許文献3】特開平1−138108号公報
【特許文献4】特開平5−311120号公報
【特許文献5】特開平10−140087号公報
【特許文献6】特開2011−79917号公報
【非特許文献】
【0011】
【非特許文献1】電子材料、1994年12月、p50
【発明の概要】
【発明が解決しようとする課題】
【0012】
従って、本発明の目的は、水蒸気等の酸化剤中における焼成工程での収縮が小さく、シリカ膜の亀裂や半導体基板との剥離が発生しにくい無機ポリシラザン及び無機ポリシラザンを含有するシリカ膜形成用塗布液を提供することにある。
【課題を解決するための手段】
【0013】
本発明者は、無機ポリシラザンの分子量、SiH3基及び窒素原子からの分岐が、焼成工程でのシリカ転化時の収縮と関係することを知見し、本発明に到達した。
【0014】
即ち、本発明は、1H―NMRスペクトルにおいて、4.75ppm以上で5.4ppm未満の範囲のピーク面積をAとし、4.5ppm以上で4.75ppm未満の範囲のピーク面積をBとし、4.2ppm以上で4.5ppm未満の範囲のピーク面積をCとしたとき、A/(B+C)の値が0.9〜1.5であり、(A+B)/Cの値が4.2〜50であり、ポリスチレン換算値による質量平均分子量が2000〜20000である、無機ポリシラザンを提供する。
また、本発明は、上記無機ポリシラザンと有機溶剤とを必須成分として含有してなるシリカ膜形成用塗布液を提供する。
さらに本発明は、上記シリカ膜形成用塗布液を基体上に塗布し、該塗布液と酸化剤とを反応させて、シリカ膜を形成することを特徴とするシリカ膜の形成方法を提供する。
【発明の効果】
【0015】
本発明によれば、酸化剤の存在下における焼成工程での収縮が小さいポリシラザンを提供することができる。
【図面の簡単な説明】
【0016】
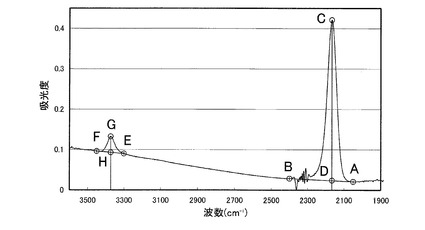
【図1】図1は、本発明におけるNH/SiH吸光度比の求め方を説明するための無機ポリシラザンの赤外スペクトルのチャートである。
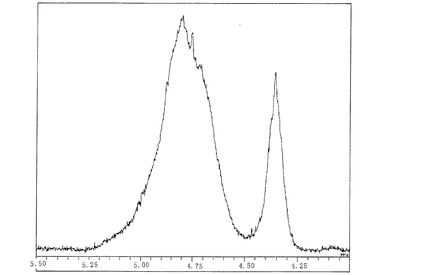
【図2】図2は、実施例1で製造したシリカ膜形成用塗布液No.1の1H―NMRスペクトルのチャートである。
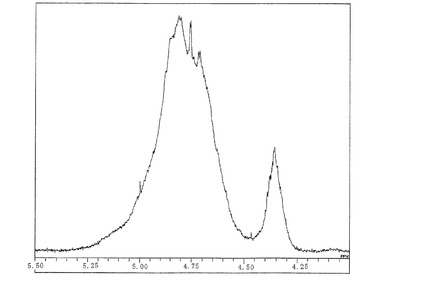
【図3】図3は、実施例2で製造したシリカ膜形成用塗布液No.2の1H―NMRスペクトルのチャートである。
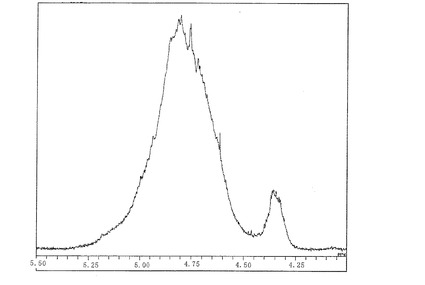
【図4】図4は、実施例3で製造したシリカ膜形成用塗布液No.3の1H―NMRスペクトルのチャートである。
【発明を実施するための形態】
【0017】
以下、本発明について好ましい実施形態に基づき詳細に説明する。
本発明の無機ポリシラザンは、1H―NMRスペクトルにおいて、4.75ppm以上で5.4ppm未満の範囲のピーク面積をAとし、4.5ppm以上で4.75ppm未満の範囲のピーク面積をBとし、4.2ppm以上で4.5ppm未満の範囲ピーク面積をCとしたとき、A/(B+C)の値が0.9〜1.5であり、(A+B)/Cの値が4.2〜50であり、ポリスチレン換算値による質量平均分子量が2000〜20000であることを特徴とする。
【0018】
無機ポリシラザンとは、−SiH2−NH−を基本ユニットとし、その構造中に有機基を持たないポリシラザンである。一般的には、直鎖のポリマーではなく、珪素原子からの分岐、窒素分子からの分岐が存在する分岐構造、架橋構造、環状構造を含むポリマーである。珪素ユニットとしては、下記式S−1〜S−4のユニットを有し、窒素ユニットとしては、下記式N−1〜N−3のユニットを有する。
【0019】
【化2】
【0020】
無機ポリシラザンの1H−NMRスペクトルにおいて、珪素原子に結合する水素原子の吸収スペクトルから、無機ポリシラザン中の上記ユニットの相対的な存在量を捕らえることが可能である。ユニットS−1の水素原子は、4.2ppm以上で4.5ppm未満の範囲に吸収を有する。ユニットS−2及びS−3の水素原子は、4.5ppm以上で5.4ppm未満の範囲に吸収を有し、ユニットS−3の水素原子の吸収は、ユニットS−2の水素原子の吸収より低磁場(高周波)域に存在する。また、ユニットN−3に含まれる珪素原子に結合する水素原子の吸収は、ユニットN−2に含まれる珪素原子に結合する水素原子の吸収よりも低磁場(高周波)域に存在する。
【0021】
ユニットS−1の水素原子の吸収は、ユニットS−1がユニットN−3に含まれる場合は、ユニットS−1がユニットN−2に含まれる場合より低磁場域に存在する。これらの吸収は、ブロードであり重なって測定される。本発明における4.2ppm以上で4.5ppm未満の範囲のピーク面積Cは、無機ポリシラザン中の−SiH3基の水素原子数に相当するものである。
【0022】
1H−NMRスペクトルにおいて、4.5ppm以上で5.4ppm未満の範囲の吸収は、低磁場側から、ユニットN−3に含まれるSiH、ユニットN−3に含まれるSiH2、ユニットN−2に含まれるSiH、及びN−2に含まれるSiH2の吸収である。
即ち、低磁場側は、ユニットN−3の珪素原子に結合する水素原子の吸収であり、高磁場側は、ユニットN−2の珪素原子に結合する水素原子の吸収である。これらのピークはブロードであり重なって測定される。低磁場側の吸収面積の割合が大きいということは、ユニットN−3の割合が多いということであり、高磁場側の吸収面積の割合が大きいということは、ユニットN−2の割合が多いということである。
ここでこの範囲の吸収を4.75ppmで区切った場合、本発明における4.75ppm以上で5.4ppm未満の範囲のピーク面積Aは、ユニットN−3の存在数が増加すれば大きくなり、4.5ppm以上で4.75ppm未満の範囲のピーク面積BはユニットN−2の存在数が増加すれば大きくなるといえる。
【0023】
即ち、本発明におけるA/(B+C)は、無機ポリシラザン中のユニットN−3の存在数の指標であり、(A+B)/Cは、無機ポリシラザン中のSiH3基の存在数の指標である。
【0024】
本発明の無機ポリシラザンにおいて、ユニットN−3の存在数の指標であるA/(B+C)の値は、0.9〜1.5であり、1.0〜1.4が好ましい。
A/(B+C)の値が0.9より小さいと、焼成工程でシリカに転化したときの収縮率の充分な低減効果が得られない。またこの値が1.5より大きくても同様である。
(A+B)/Cの値が0.9より大きいと収縮率が小さくなる理由は、ユニットN−3がシリカへ転化するときに、窒素1分子が、酸素1.5分子と置き換わることにより、ユニットが占める体積が増加することに起因すると、我々は考察している。
A/(B+C)の値が1.5より大きいと、収縮率の低減が得られない理由は、ユニットN−3が増加すると、無機ポリシラザンがシリカへ転化する場合に必要なアンモニア分子が少なくなり、その結果、無機ポリシラザン中のSi−N結合がSi−O結合に転化される割合が小さくなり、シリカに転化されなかったポリシラザン部分が、アウトガスとしてロスすることになり、ユニットN−3の収縮抑制の効果を打ち消すことになると、我々は考察している。
【0025】
本発明の無機ポリシラザンにおける(A+B)/Cの値は、4.2〜50であり、4.5〜20が好ましい。
(A+B)/Cの値が4.2より小さいと、焼成工程でシリカに転化したときの収縮率が大きくなる。また、この値が50より大きい無機ポリシラザンの製造は、困難である。(A+B)/Cの値が小さいということは、SiH3基が多いということになり、SiH3基は、シリカ転化時には、分解され、モノシランのアウトガスとしてロスすることになる。(A+B)/Cの値が50より大きい無機ポリシラザンの製造が困難な理由は、アンモニアとハロシランの反応時にハロシランの一部がポリマー化反応前に不均化反応を起こし、珪素原子に隣接する水素原子の数が変化する為であると、我々は考察している。
【0026】
本発明の無機ポリシラザンの分子量は、ポリスチレン換算値による重量平均分子量が2000〜20000であり、3000〜10000が好ましい。
重量平均分子量が2000より小さいと、シリカ膜形成時の乾燥工程や焼成工程において塗膜からのアウトガスが増え、シリカ膜の膜厚の低下や亀裂の発生が起こる。20000より大きいと、微細なパターンやアスペクト比の大きいパターンの埋め込み性が悪化し、良好なシリカ膜の形成が難しくなる。
【0027】
また、本発明の無機ポリシラザン中の低分子量成分が過剰に多い場合は、乾燥工程や焼成工程において塗膜からの揮発物又は昇華物が増え、シリカ膜の膜厚の低下や亀裂の発生が起こる場合があることから、本発明の無機ポリシラザン中の質量平均分子量が800以下である成分の割合は、40%以下であることが好ましく、30%以下であることが更に好ましい。
【0028】
尚、本発明において、質量平均分子量とは、テトラヒドロフラン(THF)を溶媒とし、示差屈折率検出器(RI検出器)を用いてGPC分析を行った場合のポリスチレン換算の質量平均分子量をいう。また、本発明の無機ポリシラザン中の質量平均分子量が800以下である成分の割合とは、GPC分析を行った場合の無機ポリシラザンのピーク面積比で、全ポリシラザン量に対する、ポリスチレン換算で質量平均分子量800以下のポリシラザン量の割合をいう。
【0029】
本発明の無機ポリシラザンの赤外スペクトルにおいて、Si−H結合に由来する吸収が2050〜2400cm-1に、N−H結合に由来する吸収が3300〜3450cm-1にある。従って、2050〜2400cm-1の吸光度は、珪素原子に結合する水素原子の数に対応し、3300〜3450cm-1の吸光度は、窒素原子に結合する水素原子の数に対応するので、赤外スペクトルにおける2050〜2400cm-1の範囲で最大の吸光度に対する3300〜3450cm-1の範囲で最大の吸光度の比は、窒素原子に結合する水素原子数/珪素原子に結合する水素原子数の指標となる。本発明では、以降この比をNH/SiH吸光度比という。
【0030】
本発明の無機ポリシラザンにおいて、NH/SiH吸光度比が0.01よりも小さいと、本発明の無機ポリシラザンの保存安定性が不良となる場合があり、0.20よりも大きいと、焼成によるシリカ転化時の収縮が大きくなる場合があることから、NH/SiH吸光度比は、0.01〜0.20であることが好ましく、0.10〜0.20であることがより好ましい。
【0031】
本発明における無機ポリシラザンの赤外スペクトルは、透過法及び反射法の何れで測定してもよい。透過法により測定する場合には、2050〜2400cm-1及び3300〜3450cm-1に、実質的に赤外スペクトルの妨害吸収のない試験片に、無機ポリシラザンを塗布した後、赤外スペクトルを測定することにより得ることができる。反射法により測定する場合も透過法と同様の試験片で測定が可能であるが、透過法に比べてS/N比が劣る場合がある。簡便で再現性が良好な方法は、例えば、両面研磨したシリコンウェハーを基体とし、スピンコーターで塗布して乾燥させた無機ポリシラザンを透過により、測定する方法である。
【0032】
上記基体上に形成される無機ポリシラザンの膜厚は、300〜1000nmの場合に精度よくNH/SiH吸光度比が得られる。赤外スペクトルの測定には、測定後のデータ処理が容易であることから、フーリエ変換型赤外分光計(FT−IR)を用いることが好ましい。
【0033】
本発明におけるNH/SiH吸光度比は、無機ポリシラザンの赤外スペクトルのスペクトルチャートから頂点強度法により得られた値である。例えば、図1において、2050cm-1、2400cm-1、3300cm-1及び3450cm-1における、吸光度曲線上の点をそれぞれ点A、点B、点E及び点Fとし、2050〜2400cm-1の範囲及び3300〜3450cm-1の範囲で吸光度が最大となる波数の、吸光度曲線上の点をそれぞれ点C及び点Gとし、点Cから基準線(吸光度0となる線、ブランク)への垂線と線ABとの交点を点D、点Gから基準線への垂線と線EFとの交点を点Hとするとき、NH/SiH吸光度比は、線分CDに対する線分GHの比に相当する。即ち、本発明のNH/SiH吸光度比は、無機ポリシラザンの赤外スペクトルのスペクトルチャートにおいて、2050cm-1の吸光度の点と2400cm-1の吸光度の点を結ぶ線をベースラインとした2050〜2400cm-1の吸光度最大値に対する、3300cm-1の吸光度の点と3450cm-1の吸光度の点を結ぶ線をベースライン3300〜3450cm-1の吸光度最大値の比である。
尚、通常、無機ポリシラザンは、2050〜2400cm-1の範囲で吸光度が最大となるのは2166cm-1付近であり、3300〜3450cm-1の範囲で吸光度が最大となるのは3377cm-1付近である。
【0034】
本発明の無機ポリシラザンは、波長633nmにおける屈折率が、1.550よりも小さい場合には、焼成によるシリカ転化時の収縮が大きくなる場合があり、1.650よりも大きい場合には、本発明のシリカ膜形成用塗布液の保存安定性が不良となる場合があるので、波長633nmにおける屈折率は、1.550〜1.650であることが好ましく、1.560〜1.640であることが更に好ましく、1.570〜1.630であることが最も好ましい。
【0035】
上記の屈折率の測定方法は、例えば、無機ポリシラザン又は無機ポリシラザンを溶解又は分散せしめた組成物を基体上に、スピンコート法、ディップコート法、ナイフコート法、ロールコート法等の方法により塗布し、乾燥して無機ポリシラザン膜を形成し測定すればよい。乾燥は、無機ポリシラザン膜の膜厚によって異なるが、500〜1000nmの場合には、150℃で1分以上、好ましくは150℃で3分程度加熱することが好ましい。ケイ素含量に対する窒素含量の比が同一である無機ポリシラザンの場合、屈折率の高い無機ポリシラザンの方が、水素含量が少なく、分子中に多数の環構造を有しており、これが、シリカ膜形成用塗布液の保存安定性や水蒸気中における焼成工程での収縮に影響を与えているものと考えられる。
【0036】
本発明の無機ポリシラザンの製造方法は、特に制限されず周知の無機ポリシラザンの製造方法を適用又は応用して製造すればよい。例えば、ハロシラン化合物とアンモニアとを直接反応させてもよく、ハロシラン化合物に塩基等の付加物を付加させた付加体を形成し、その付加体とアンモニアとを反応させてもよい。このような付加体を経由する無機ポリシラザンの製造方法は、例えば、特開昭60−145903号公報、特開昭61−174108号公報等に開示されている。
本発明の無機ポリシラザンの製造方法としては、反応が制御できる点から、ハロシラン化合物と塩基とを反応させて付加体を形成した後、該付加体とアンモニアとを反応させる方法が好ましい。
【0037】
ハロシラン化合物と塩基とを反応させて付加体を形成した後、該付加体とアンモニアとを反応させる無機ポリシラザンの製造方法において、当該付加体とアンモニアとの反応は、通常、−50〜20℃、好ましくは−10〜15℃の温度で行う。
【0038】
本発明の無機ポリシラザンの原料に使用するハロシラン化合物としては、ジクロロシラン、ジブロモシラン、クロロブロモシラン等のジハロシラン化合物;トリクロロシラン、トリブロモシラン、ジクロロブロモシラン、クロロジブロモシラン等のトリハロシラン化合物、テトラクロロシラン、テトラブロモシランが挙げられるが、ハロシランとしては、クロロシラン類が安価であるので好ましい。ハロシラン化合物は1種のみを使用してもよいし、2種以上を組み合わせて使用してもよい。ジハロシラン化合物を使用した無機ポリシラザンは成膜性に優れ、トリハロシラン化合物を使用した無機ポリシラザンは焼結時の収縮が少ないという利点があり、本発明の無機ポリシラザンを製造する場合には、ジハロシラン化合物、トリハロシラン化合物、又はジハロシラン化合物とトリハロシラン化合物を混合して使用することが好ましい。
【0039】
ジハロシラン化合物とトリハロシラン化合物を混合して使用する場合、ユニットS−2の数を制御する点から、その割合が、ジハロシラン化合物1モルに対してトリハロシラン化合物が、0.01〜2モルであることが好ましく、0.03〜1モルであることが更に好ましく、0.05〜0.5モルであることが最も好ましい。
【0040】
また、付加体を形成するための付加物である塩基は、ハロシラン化合物との付加体を形成する反応以外は不活性である塩基がよい。このような塩基としては、例えば、トリメチルアミン、トリエチルアミン、トリブチルアミン、ジメチルアニリン等の3級アミン類;ピリジン、ピコリン等のピリジン類が挙げられ、工業的な入手の容易さと取扱いの容易さの点から、ピリジン及びピコリンが好ましく、ピリジンが更に好ましい。使用する塩基の量は、ハロシラン化合物のハロゲン原子に対して、1倍モル以上であればよいが、付加体の形成が不十分とならないよう、1.1倍モル以上であることが好ましい。
【0041】
本発明の無機ポリシラザンの製造方法において、上記付加体が形成されると、反応系の流動性が低下するので、付加体の形成反応は有機溶剤中で行うことが好ましい。当該溶剤は、無機ポリシラザンと反応しない有機溶剤を使用することができる。例えば、ペンタン、ヘキサン、ヘプタン、オクタン、2,2,4−トリメチルペンタン(イソオクタンともいう)、イソノナン、2,2,4,6,6−ペンタメチルヘプタン(イソドデカンともいう)等の飽和鎖状炭化水素化合物;シクロペンタン、シクロヘキサン、メチルシクロヘキサン、エチルシクロヘキサン、デカリン等の飽和環状炭化水素化合物;ベンゼン、トルエン、キシレン、エチルベンゼン、クメン、プソイドクメン、テトラリン等の芳香族炭化水素化合物;ジエチルエーテル、ジプロピルエーテル、ジイソプロピルエーテル、ジブチルエーテル、ジイソブチルエーテル、t−ブチルメチルエーテル、テトラヒドロフラン、ジオキサン、1,2−ジメトキシエタン等のエーテル化合物等が挙げられる。
【0042】
また、有機溶剤の代わりの反応溶媒として、付加物である塩基を過剰量用いて、過剰量の塩基を溶媒としてもよい。特に好ましいのは、ピリジンを付加物として、これを形成反応が終了しても流動性を保てる程度に過剰量使用し、他の有機溶剤を使用しないことである。この場合、ピリジンの使用量は、ハロシラン化合物に対して3〜30倍モルであることが好ましく、4〜25倍モルであることが更に好ましく、5〜20倍モルであることが好ましい。また、付加体形成による流動性の低下を防ぐため、有機溶剤、付加物、有機溶剤と付加物を含有する混合溶媒に、ハロシラン化合物とアンモニアとを分割して仕込んでもよく、又は連続的に同時に仕込んでもよい。
【0043】
付加体を経由する製造方法においては、アンモニアの使用量は、化学量論上、反応に使用するハロシラン化合物のハロゲン原子に対して等モル以上(1倍モル以上)であればよいが、反応を完結させるのに十分であり経済性を考慮すると、アンモニアの使用量は、反応に使用するハロシラン化合物のハロゲン原子に対して、1.0〜3.0倍モルであることが好ましく、1.1〜2.5倍モルであることが更に好ましく、1.2〜2.0倍モルであることが最も好ましい。
【0044】
アンモニアとの反応後は、必要に応じて過剰のアンモニアを除去し、生成したハロゲン化アンモニウムを濾過等により除去する。その後、必要に応じて公知の方法により、所望の有機溶剤に溶剤置換等を行えばよい。
【0045】
また、本発明の無機ポリシラザンは、生成した塩の除去前、又は除去後に、無機ポリシラザン分子中のSiH基とNH基とを反応させてSi−N結合を生成させることにより、分子内反応による環状化、分子間反応による高分子量化等を行ってもよく、これにより、SiH3基の減少、質量平均分子量の増加、質量平均分子量が800以下である成分の減少、NH/SiH吸光度比の増加、屈折率の増加等の調整を図ってもよい。無機ポリシラザンのSiH基とNH基を反応させてSi−N結合を生成させる方法としては、例えば、ピリジン、ピコリン等の塩基性溶媒中で加熱する方法(例えば、特開平1−138108号公報を参照)、アルカリ金属水素化物、アルカリ金属アルコキシド、無水アルカリ金属水酸化物等のアルカリ金属含有塩基性触媒による方法(例えば、特開昭60−226890号公報を参照)、テトラメチルアンモニウムヒドロキシド等の4級アンモニウム化合物を触媒とする方法(例えば、特開平5−170914号公報を参照)、硝酸アンモニウム、酢酸アンモニウム等の酸触媒を使用する方法(例えば、特開表2003−514822号公報を参照)等が挙げられ、反応に使用した付加物又は当該付加物を含有する溶媒中で加熱する方法が好ましい。
【0046】
付加体を経由する方法で無機ポリシラザンを製造する場合には、付加体(例えばジクロロシランとピリジン)とアンモニアとが反応して付加物(例えばピリジン)が遊離することから、遊離した付加物を塩基性溶媒として使用することが可能である。従って、付加体を経由して無機ポリシラザンを製造した後、遊離した付加物を溶媒として加熱することにより無機ポリシラザンのSiH基とNH基を反応させてSi−N結合を生成させることは、原料の有効利用及び製造工程の簡略化の点から好ましい。
【0047】
本発明のシリカ膜形成用塗布剤は、上記の本発明の無機ポリシラザンと、有機溶剤を必須成分として含有する組成物であり、基体に塗布しやすい濃度に調製される。
【0048】
本発明のシリカ膜形成用塗布剤に使用される有機溶剤は、無機ポリシラザンと反応することで塗布性を損ねる程の変質や反応をきたすものでなければ、特に限定されない。水酸基、アルデヒド基、ケトン基、カルボキシル基、エステル基等は、無機ポリシラザンとの高い反応性を有することから、これらの基を有しないものが好ましい。本発明のシリカ膜形成用塗布液の好ましい有機溶剤としては、例えば、ペンタン、ヘキサン、ヘプタン、オクタン、2,2,4−トリメチルペンタン(イソオクタンともいう)、イソノナン、2,2,4,6,6−ペンタメチルヘプタン(イソドデカンともいう)等の飽和鎖状炭化水素化合物;シクロペンタン、シクロヘキサン、メチルシクロヘキサン、デカリン等の飽和環状炭化水素化合物;ベンゼン、トルエン、キシレン、エチルベンゼン、クメン、プソイドクメン、テトラリン等の芳香族炭化水素化合物;ジエチルエーテル、ジプロピルエーテル、ジイソプロピルエーテル、ジブチルエーテル、ジイソブチルエーテル、t−ブチルメチルエーテル、テトラヒドロフラン、ジオキサン、1,2−ジメトキシエタン等のエーテル化合物等が挙げられ、中でも、塗布性が良好であることから、キシレン、ジブチルエーテルが好ましく、保存安定性が良好であることからジブチルエーテルが更に好ましい。有機溶剤は、1種類のみでもよいが、蒸発速度の調整等の目的で2種以上を組み合わせて用いてもよい。
【0049】
エーテル化合物は、その原料、製造工程の副生成物、保存中の劣化生成物として、アルコール化合物、アルデヒド化合物、ケトン化合物、カルボン酸化合物、エステル化合物等が含まれることがある。本発明のシリカ膜形成用塗布液の有機溶剤に、これらの化合物が含まれる場合には焼成工程での収縮が大きくなる場合があることから、無機ポリシラザンと混合する以前に、これらのアルコール化合物、アルデヒド化合物、ケトン化合物、カルボン酸化合物及びエステル化合物の含量の合計を、ジブチルエーテルに対して0.1質量%以下にしておくことが好ましく、0.05質量%以下であることが更に好ましく、0.01質量%以下であることが最も好ましい。
【0050】
本発明のシリカ膜形成用塗布液中の無機ポリシラザンの含量が、あまりに低い場合には、シリカ膜の成膜性が不十分となり、あまりに高い場合には本発明のシリカ膜形成用塗布液の保存安定性が不十分となりゲル物が発生する場合があることから、無機ポリシラザンの含量は、1〜40質量%が好ましく、3〜35質量%が更に好ましく、5〜30質量%が最も好ましい。
【0051】
本発明のシリカ膜形成用塗布液は、主として、該塗布液を基体(対象材料)上に塗布し、該塗布液と、酸化剤とを反応させて、形成されるシリカ膜として、従来、無機ポリシラザンが使用されてきた用途、例えば、半導体装置の絶縁膜、フラットパネルディスプレイの保護膜、光学関連製品の反射防止膜等に使用でき、特に半導体装置の絶縁膜に好適に使用できる。
【0052】
例えば、半導体装置の絶縁膜を形成する場合には、本発明のシリカ形成用塗布液を対象材料(基体)上に塗布し、塗膜を形成する塗布工程、塗膜から有機溶媒を除去する乾燥工程、水蒸気中において焼成しシリカ膜を形成する焼成工程を含む製造方法が好ましい。
本発明のシリカ膜形成用塗布液を対象材料に塗布する場合には、特に限定されず、スプレー法、スピンコート法、ディップコート法、ロールコート法、フローコート法、スクリーン印刷法、転写印刷法等のいずれの塗布方法でもよいが、膜厚が薄く均一な塗膜が形成できることからスピンコート法が好ましい。
【0053】
乾燥工程の乾燥温度及び時間は、使用する有機溶媒及び塗膜の膜厚により異なるが、80〜200℃、好ましくは120〜170℃において、1〜30分、好ましくは2〜10分加熱することが好ましい。乾燥雰囲気は、酸素中、空気中、不活性ガス中の何れでもよい。また、焼成工程は、相対湿度20〜100%の水蒸気雰囲気下に200〜1200℃の温度が好適な範囲である。焼成温度が低い場合には、反応が十分進行しない場合があるとともに、シラノール基の残存による絶縁性の低下の懸念があり、焼成温度が高いと製造コストの問題があることから、水蒸気雰囲気下の焼成の温度は、300〜1000℃が好ましく、700〜900℃が更に好ましい。焼成する場合は、700℃以上の温度により1段階で焼成してもよいし、200〜500℃、好ましくは300〜450℃で30〜60分焼成した後に、450〜1200℃、好ましくは600〜1000℃、更に好ましくは700〜900℃で焼成する2段階の焼成でもよい。シリカ膜の収縮が少なく、亀裂が生じにくいことから2段階の焼成が好ましい。このほか、200〜500℃、好ましくは350〜450℃で30〜60分焼成した後に、20〜80℃の蒸留水に浸漬させる低温焼成法(例えば、特開平7−223867号公報を参照)でもよいが、低温焼成法ではシラノール基の残存による絶縁性の低下が起こることから、低温焼成後に大気中で700〜900℃で5〜60分程度加熱することが好ましい。
【実施例】
【0054】
以下、実施例により本発明を具体的に説明するが、これらは本発明の範囲を限定するものではない。尚、実施例中の「部」や「%」は質量によるものである。また、溶媒に用いたジブチルエーテルは純度が99.99%であり、アルコール化合物、アルデヒド化合物、ケトン化合物、カルボン酸化合物及びエステル化合物の合計の含量が0.01%以下であるものを用いた。
【0055】
[実施例1]
攪拌機、温度計及び導入管を備えた3000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン2310g(29.2モル)を仕込み、撹拌しながら、トリクロロシラン48.6g(0.36モル)とジクロロシラン82.6g(0.82モル)を反応温度0〜5℃となるように冷却しながら、それぞれ1時間かけて滴下し、ピリジン付加体を生成させた。アンモニア78.9g(4.64モル)を、反応温度が10℃を超えないように冷却しながら3時間かけて導入管からフィードし、窒素ガスを吹き込みながら、更に10℃で1.5時間撹拌を行い、反応を完結させた。この反応液を10℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾別し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに溶媒交換をした。得られた溶液を、120℃で6時間加熱した後、濾過径0.1μmのPTFE製カートリッジフィルターで濾過を行い、無機ポリシラザン含量が11.3%であるシリカ膜形成用塗布液No.1を得た。
【0056】
[実施例2]
攪拌機、温度計及び導入管を備えた3000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン2310g(29.2モル)を仕込み、撹拌しながら、トリクロロシラン50.4g(0.37モル)とジクロロシラン82.9g(0.82モル)を、反応温度が−10〜0℃となるように冷却しながら、それぞれ1時間かけて滴下し、ピリジン付加体を生成させた。アンモニア78.9g(4.61モル)を、反応温度が5℃を超えないように冷却しながら3時間かけて導入管からフィードし、窒素ガスを吹き込みながら、更に10℃で1.5時間撹拌を行い、反応を完結させた。この反応液を10℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾別し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに溶媒交換をした。得られた溶液を、120℃で6時間加熱した後、濾過径0.1μmのPTFE製カートリッジフィルターで濾過を行い、無機ポリシラザン含量が18.7%であるシリカ膜形成用塗布液No.2を得た。
【0057】
[実施例3]
攪拌機、温度計及び導入管を備えた3000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン2411g(30.5モル)を仕込み、撹拌しながら、トリクロロシラン69.8g(0.52モル)とジクロロシラン51.3g(0.51モル)を、反応温度が−10〜0℃となるように冷却しながら、それぞれ1時間かけて滴下し、ピリジン付加体を生成させた。アンモニア74.4g(4.35モル)を、反応温度−10〜0℃で3時間かけて導入管からフィードし、窒素ガスを吹き込みながら、更に10℃で1.5時間撹拌を行い、反応を完結させた。この反応液を10℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾別し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに溶媒交換をした。得られた溶液を、120℃で6時間加熱した後、濾過径0.1μmのPTFE製カートリッジフィルターで濾過を行い、無機ポリシラザン含量が9.64%であるシリカ膜形成用塗布液No.3を得た。
【0058】
[比較例1]
攪拌機、温度計及び導入管を備えた3000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン1646g(20.8モル)を仕込み、撹拌しながら、ジクロロシラン310g(3.1モル)を、反応温度0〜5℃で1時間かけて導入管からフィードし、ジクロロシランのピリジンアダクツを生成させた。アンモニア180g(10.6モル)を、反応温度0〜5℃で1時間かけて導入管からフィードし、更に10℃で1.5時間撹拌を行い、反応を完結させた。この反応液を10℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾過し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに溶媒交換をした。得られた溶液を、窒素雰囲気下、濾過径0.1μmのPTFE製カートリッジフィルターにて濾過を行い、無機ポリシラザン含量が19.0%である比較用塗布液1を得た。
【0059】
[比較例2]
攪拌機、温度計及び導入管を備えた3000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン2248g(28.4モル)を仕込み、撹拌しながら、ジクロロシラン191.0g(1.89モル)とアンモニア113.0g(6.65モル)を、反応温度が0〜5℃となるように冷却しながら、それぞれ3時間かけてフィードし、窒素ガスを吹き込みながら、更に10℃で1.5時間撹拌を行い、反応を完結させた。この反応液を10℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾別し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに溶媒交換をした。得られた溶液を、120℃で6時間加熱した後、濾過径0.1μmのPTFE製カートリッジフィルターで濾過を行い、無機ポリシラザン含量が19.2%である比較用塗布液2を得た。
【0060】
[比較例3]
攪拌機、温度計及び導入管を備えた3000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン2044g(25.8モル)を仕込み、撹拌しながら、ジクロロシラン174.0g(1.72モル)とアンモニア103.0g(6.06モル)を、反応温度が0〜5℃となるように冷却しながら、それぞれ3時間かけてフィードし、窒素ガスを吹き込みながら、更に10℃で1.5時間撹拌を行い、反応を完結させた。この反応液を10℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾別し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに溶媒交換をした。得られた溶液を、120℃で6時間加熱した後、濾過径0.1μmのPTFE製カートリッジフィルターで濾過を行い、無機ポリシラザン含量が19.3%である比較用塗布液3を得た。
【0061】
[比較例4]
攪拌機、温度計及び導入管を備えた3000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン2303g(29.1モル)を仕込み、撹拌しながら、ジクロロシラン280.0g(2.77モル)とアンモニア165.0g(9.71モル)を、反応温度が−10〜0℃となるように冷却しながら、それぞれ4時間かけてフィードし、窒素ガスを吹き込みながら、更に0℃で1.5時間撹拌を行い、反応を完結させた。この反応液を10℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾別し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに溶媒交換をした。得られた溶液を、120℃で6時間加熱した後、濾過径0.1μmのPTFE製カートリッジフィルターで濾過を行い、無機ポリシラザン含量が19.0%である比較用塗布液4を得た。
【0062】
[比較例5]
攪拌機、温度計及び導入管を備えた3000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン2044g(25.8モル)を仕込み、撹拌しながら、ジクロロシラン325.7g(3.22モル)とアンモニア192.1g(11.3モル)を、反応温度が−10〜0℃となるように冷却しながら、それぞれ2時間かけてフィードし、窒素ガスを吹き込みながら、更に0℃で1.5時間撹拌を行い、反応を完結させた。この反応液を10℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾別し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに溶媒交換をした。得られた溶液を、120℃で6時間加熱した後、濾過径0.1μmのPTFE製カートリッジフィルターで濾過を行い、無機ポリシラザン含量が19.2%である比較用塗布液5を得た。
【0063】
[比較例6]
攪拌機、温度計及び導入管を備えた3000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン2044g(25.8モル)を仕込み、撹拌しながら、ジクロロシラン260.6g(2.58モル)とアンモニア131.6g(7.74モル)を、反応温度が−10〜0℃となるように冷却しながら、それぞれ1.5時間かけてフィードし、窒素ガスを吹き込みながら、更に0℃で1.5時間撹拌を行い、反応を完結させた。この反応液を10℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾別し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに溶媒交換をした。得られた溶液を、120℃で6時間加熱した後、濾過径0.1μmのPTFE製カートリッジフィルターで濾過を行い、無機ポリシラザン含量が20.3%である比較用塗布液6を得た。
【0064】
[比較例7]
攪拌機、温度計及び導入管を備えた5000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン4300g(54.4モル)を仕込み、撹拌しながら、ジクロロシラン545g(5.4モル)を、反応温度−40〜−30℃で1時間かけて導入管からフィードし、ジクロロシランのピリジンアダクツを生成させた。アンモニア325g(19.1モル)を、反応温度−40〜−30℃で1時間かけて導入管からフィードし、更に−20〜−15℃で2時間撹拌を行い、反応を完結させた。この反応液を25℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾過し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに常法により交換し、更にアルゴンガス雰囲気中で、濾過径0.1μmのPTFE製カートリッジフィルターにて濾過を行い、無機ポリシラザン含量が19.0%である比較用塗布液7を得た。
【0065】
[比較例8]
比較例7において、アンモニアの反応温度を−40〜−30℃から−15〜−12℃に変更し、その後−15〜−12℃で2時間撹拌した以外は、比較例7と同様の操作を行い、無機ポリシラザン含量が19.1%である比較用塗布液8を得た。
【0066】
[比較例9]
比較例7において、ジクロロシラン545g(5.4モル)の代わりにジクロロシラン444g(4.4モル)とトリクロロシラン13.6g(1.0モル)との混合物を使用し、アンモニアを325g(19.1モル)から340g(20.0モル)に増やした以外は、比較例7と同様の操作を行い、無機ポリシラザン含量が19.2%である比較用塗布液9を得た。
【0067】
<分析:1H―NMR分析>
実施例1〜3で得たシリカ膜形成用塗布液No.1〜3及び比較例1〜9で得た比較用塗布液1〜9について、1H―NMRを測定した。シリカ膜形成用塗布液No.1〜No.4のチャートを図2〜図4に示す。1H―NMRスペクトルにおいて、4.75ppm以上で5.4ppm未満の範囲のピーク面積をAとし、4.5ppm以上で4.75ppm未満の範囲のピーク面積をB、4.2ppm以上で4.5ppm未満の範囲のピーク面積をCとして、A/(B+C)の値、(A+B)/Cの値を算出した。結果を〔表1〕に示す。
【0068】
<分析:GPC>
実施例1〜3で得たシリカ膜形成用塗布液No.1〜3及び比較例1〜9で得た比較用塗布液1〜9について、GPCの結果から、無機ポリシラザンの質量平均分子量、及び質量平均分子量800以下の成分の含量をそれぞれ算出した。結果を〔表1〕に示す。カラムは東ソー(株)製、スーパーマルチポアHZ−Mを使用した。
【0069】
<無機ポリシラザン塗膜の分析:膜厚、IR分析>
実施例1〜3で得たシリカ膜形成用塗布液No.1〜3及び比較例1〜9で得た比較用塗布液1〜9を、両面を研磨した厚さ4インチのシリコンウェハーに、乾燥後の無機シラザンの膜厚が580〜620nmとなるようにスピンコート法により塗布してから150℃で3分間乾燥して、無機ポリシラザンの塗膜を有するシリコンウェハーを調製し、塗膜の膜厚、FT−IRを測定した。なお、FT−IR測定では、両面を研磨したシリコンウェハーをリファレンスとした。また、膜厚は、Filmetrics社製、F−20を用いて測定した。膜厚及びFT−IRの結果から算出したNH/SiH吸光度比を〔表1〕に示す。
【0070】
【表1】
【0071】
[実施例4及び比較例10]
上記の無機ポリシラザンの塗膜の分析に用いたシリコンウェハーを用いて、1段目の焼成として、相対湿度90%で温度300℃のオーブンに30分、2段目の焼成として、相対湿度10%で温度900℃のオーブンで30分、焼成することによりシリカ絶縁膜を形成させ、シリカ膜の膜厚を測定した。乾燥後の無機シラザンの膜厚に対するシリカ絶縁膜の膜厚を硬化収縮率(%)とした。結果を〔表2〕に示す。
【0072】
【表2】
【0073】
上記〔表1〕及び〔表2〕の結果から、A/(B+C)の値、(A+B)/Cの値及び質量平均分子量が所定の範囲内にある、本願発明の無機ポリシラザンを含有するシリカ膜形成用塗布液は、A/(B+C)の値、(A+B)/Cの値及び質量平均分子量が所定の範囲内にない無機ポリシラザンを含有する比較用塗布液に比して、硬化収縮率が小さく、シリカ膜の亀裂や半導体基板との剥離が発生しにくいことが明らかである。
【技術分野】
【0001】
本発明は、特定の構成の無機ポリシラザン、当該無機ポリシラザンと有機溶剤とを必須成分として含有してなるシリカ膜形成用塗布液及びこれを用いたシリカ膜の形成方法に関する。
【背景技術】
【0002】
酸化珪素を主成分とするシリカ膜は、絶縁性、耐熱性、耐摩耗性及び耐蝕性の点で優れていることから、ハードコート材及び半導体装置の絶縁膜として広く使用されている。半導体装置の微細化に伴い、狭い隙間を埋め込む絶縁膜材料が望まれている。半導体装置に用いられる絶縁膜は、例えばCVD(Chemical Vapor Deposition、化学気相成長)法や塗布法により形成される。塗布法は、コストと生産性の面で優れていることから、品質の向上を目指し種々の材料が検討されている。
【0003】
ポリシラザンは、−SiH2−NH−を基本ユニットとする高分子化合物であり、比較的安価な方法である塗布法により、狭い隙間にも品質のよい酸化珪素を主成分とするシリカ膜を形成することができる。
【0004】
ポリシラザンを用いてシリカ膜を形成する方法としては、(1)ポリシラザンのキシレンやジブチルエーテル等の溶液を半導体基板等にスピンコート等により塗布する塗布工程、(2)ポリシラザンが塗布された半導体基板等を150℃程度に加熱し溶媒を蒸発させる乾燥工程、(3)この半導体基板等を水蒸気等の酸化剤の存在下において230〜900℃程度で焼成する焼成工程による方法(例えば、特許文献1及び2を参照)が知られている。ポリシラザンは、水蒸気における焼成工程でシリカに転化される。
尚、焼成工程において、ポリシラザンが、酸化剤である水蒸気によりシリカに転化される反応は、下記反応式(1)及び反応式(2)で表されることが知られている(例えば、非特許文献1参照)。
【化1】
【0005】
ポリシラザンを用いたシリカ膜の形成では、ポリシラザン塗膜がシリカ膜に変化する過程で収縮が起こる。ポリシラザンからシリカへの反応性を高めるとともに、シリカ表面のシラノール基(Si−OH)を減少させ絶縁性を向上させるためには、水蒸気中における焼成工程をより高温で行うことが好ましいが、高温における焼成は、このような収縮も大きくなる。水蒸気中における焼成工程での収縮率が高い場合には、シリカ膜の亀裂や、シリカ膜の半導体基板からの剥離が発生する場合があり、特に、半導体装置の素子間隔が狭い隙間を埋め込む素子間分離用途に無機ポリシラザンを使用し、高温で焼成する場合に、亀裂や剥離が発生しやすいという問題があった。今後、半導体素子の間隔を、更に狭めた半導体装置が要求されていることから、収縮の抑制された無機ポリシラザンが求められている。
【0006】
特許文献3には、一分子中のSiH3基に対するSiH2基の比が2.5〜8.4であり、元素比率でSi:N:H=50〜70質量%:20〜34質量%:5〜9質量%であるポリシラザンが耐熱性、耐摩耗性及び耐薬品性に優れると共に、表面硬度の高い被膜を与え、セラミックス成形体、特にセラミックス成形焼結体用のバインダーとして好適に使用できることが開示されている。しかしながら、このようなポリシラザンは、SiH3基の含量が多いために水蒸気中における焼成工程での収縮が大きく、500℃以上で焼成する場合にはシリカ膜の亀裂が発生しやすくなるという問題があった。
【0007】
特許文献4には、1H―NMRスペクトルのピーク面積比における、SiH1、SiH2及びSiH3の和に対するSiH3の割合が0.13〜0.45であり、数平均分子量200〜100,000であるポリシラザンを必須成分とする紫外線遮蔽ガラスの保護膜形成用組成物を、ガラス平面上の紫外線遮蔽層に塗布し、乾燥空気中で加熱することにより力学的強度や化学的安定に優れた保護膜が形成されることが開示されている。
【0008】
特許文献5には、1H―NMRスペクトルのピーク面積比における、SiH1とSiH2の和に対するSiH3の割合を0.15〜0.45に調整したポリシラザンの不活性有機溶剤溶液から成る層間絶縁膜形成用塗布液が、保存安定性及び塗布特性に優れると共に、絶縁性が高く、ち密で表面形状の良好な被膜を再現性よく形成することができることが開示されている。また、ポリシラザンの活性水素の一部をトリメチルシリル基に置換することにより調整が可能であり、調整剤としてヘキサメチルジシラザンを使用することが開示されている。しかしながら、ヘキサメチルジシラザンを反応させたポリシラザンは、水蒸気中における焼成工程での収縮が大きく、500℃以上で焼成する場合には、シリカ膜の亀裂が発生しやすくなるという問題があった。
【0009】
特許文献6には、1H−NMRスペクトルにおいて、SiH3基に由来する4.3〜4.5ppmのピーク面積に対する、SiH1基とSiH2基とに由来する4.5〜5.3ppmのピーク面積の比が、4.2〜50である無機ポリシラザンと、有機溶媒とを含有することを特徴とする絶縁膜形成用塗布液が、水蒸気中における焼成工程での収縮が小さく、シリカ被膜の亀裂や半導体基板との剥離が発生しにくい絶縁膜形成用塗布液、それを用いた絶縁膜及びそれに用いる化合物の製造方法を提供することが開示されている。しかしながら、シリカ膜中の残留カーボン低減のためには、高温焼成が求められる場合があり、更なる熱収縮改善の要求がある。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特開平7−223867号公報
【特許文献2】特開2001−308090号公報
【特許文献3】特開平1−138108号公報
【特許文献4】特開平5−311120号公報
【特許文献5】特開平10−140087号公報
【特許文献6】特開2011−79917号公報
【非特許文献】
【0011】
【非特許文献1】電子材料、1994年12月、p50
【発明の概要】
【発明が解決しようとする課題】
【0012】
従って、本発明の目的は、水蒸気等の酸化剤中における焼成工程での収縮が小さく、シリカ膜の亀裂や半導体基板との剥離が発生しにくい無機ポリシラザン及び無機ポリシラザンを含有するシリカ膜形成用塗布液を提供することにある。
【課題を解決するための手段】
【0013】
本発明者は、無機ポリシラザンの分子量、SiH3基及び窒素原子からの分岐が、焼成工程でのシリカ転化時の収縮と関係することを知見し、本発明に到達した。
【0014】
即ち、本発明は、1H―NMRスペクトルにおいて、4.75ppm以上で5.4ppm未満の範囲のピーク面積をAとし、4.5ppm以上で4.75ppm未満の範囲のピーク面積をBとし、4.2ppm以上で4.5ppm未満の範囲のピーク面積をCとしたとき、A/(B+C)の値が0.9〜1.5であり、(A+B)/Cの値が4.2〜50であり、ポリスチレン換算値による質量平均分子量が2000〜20000である、無機ポリシラザンを提供する。
また、本発明は、上記無機ポリシラザンと有機溶剤とを必須成分として含有してなるシリカ膜形成用塗布液を提供する。
さらに本発明は、上記シリカ膜形成用塗布液を基体上に塗布し、該塗布液と酸化剤とを反応させて、シリカ膜を形成することを特徴とするシリカ膜の形成方法を提供する。
【発明の効果】
【0015】
本発明によれば、酸化剤の存在下における焼成工程での収縮が小さいポリシラザンを提供することができる。
【図面の簡単な説明】
【0016】
【図1】図1は、本発明におけるNH/SiH吸光度比の求め方を説明するための無機ポリシラザンの赤外スペクトルのチャートである。
【図2】図2は、実施例1で製造したシリカ膜形成用塗布液No.1の1H―NMRスペクトルのチャートである。
【図3】図3は、実施例2で製造したシリカ膜形成用塗布液No.2の1H―NMRスペクトルのチャートである。
【図4】図4は、実施例3で製造したシリカ膜形成用塗布液No.3の1H―NMRスペクトルのチャートである。
【発明を実施するための形態】
【0017】
以下、本発明について好ましい実施形態に基づき詳細に説明する。
本発明の無機ポリシラザンは、1H―NMRスペクトルにおいて、4.75ppm以上で5.4ppm未満の範囲のピーク面積をAとし、4.5ppm以上で4.75ppm未満の範囲のピーク面積をBとし、4.2ppm以上で4.5ppm未満の範囲ピーク面積をCとしたとき、A/(B+C)の値が0.9〜1.5であり、(A+B)/Cの値が4.2〜50であり、ポリスチレン換算値による質量平均分子量が2000〜20000であることを特徴とする。
【0018】
無機ポリシラザンとは、−SiH2−NH−を基本ユニットとし、その構造中に有機基を持たないポリシラザンである。一般的には、直鎖のポリマーではなく、珪素原子からの分岐、窒素分子からの分岐が存在する分岐構造、架橋構造、環状構造を含むポリマーである。珪素ユニットとしては、下記式S−1〜S−4のユニットを有し、窒素ユニットとしては、下記式N−1〜N−3のユニットを有する。
【0019】
【化2】
【0020】
無機ポリシラザンの1H−NMRスペクトルにおいて、珪素原子に結合する水素原子の吸収スペクトルから、無機ポリシラザン中の上記ユニットの相対的な存在量を捕らえることが可能である。ユニットS−1の水素原子は、4.2ppm以上で4.5ppm未満の範囲に吸収を有する。ユニットS−2及びS−3の水素原子は、4.5ppm以上で5.4ppm未満の範囲に吸収を有し、ユニットS−3の水素原子の吸収は、ユニットS−2の水素原子の吸収より低磁場(高周波)域に存在する。また、ユニットN−3に含まれる珪素原子に結合する水素原子の吸収は、ユニットN−2に含まれる珪素原子に結合する水素原子の吸収よりも低磁場(高周波)域に存在する。
【0021】
ユニットS−1の水素原子の吸収は、ユニットS−1がユニットN−3に含まれる場合は、ユニットS−1がユニットN−2に含まれる場合より低磁場域に存在する。これらの吸収は、ブロードであり重なって測定される。本発明における4.2ppm以上で4.5ppm未満の範囲のピーク面積Cは、無機ポリシラザン中の−SiH3基の水素原子数に相当するものである。
【0022】
1H−NMRスペクトルにおいて、4.5ppm以上で5.4ppm未満の範囲の吸収は、低磁場側から、ユニットN−3に含まれるSiH、ユニットN−3に含まれるSiH2、ユニットN−2に含まれるSiH、及びN−2に含まれるSiH2の吸収である。
即ち、低磁場側は、ユニットN−3の珪素原子に結合する水素原子の吸収であり、高磁場側は、ユニットN−2の珪素原子に結合する水素原子の吸収である。これらのピークはブロードであり重なって測定される。低磁場側の吸収面積の割合が大きいということは、ユニットN−3の割合が多いということであり、高磁場側の吸収面積の割合が大きいということは、ユニットN−2の割合が多いということである。
ここでこの範囲の吸収を4.75ppmで区切った場合、本発明における4.75ppm以上で5.4ppm未満の範囲のピーク面積Aは、ユニットN−3の存在数が増加すれば大きくなり、4.5ppm以上で4.75ppm未満の範囲のピーク面積BはユニットN−2の存在数が増加すれば大きくなるといえる。
【0023】
即ち、本発明におけるA/(B+C)は、無機ポリシラザン中のユニットN−3の存在数の指標であり、(A+B)/Cは、無機ポリシラザン中のSiH3基の存在数の指標である。
【0024】
本発明の無機ポリシラザンにおいて、ユニットN−3の存在数の指標であるA/(B+C)の値は、0.9〜1.5であり、1.0〜1.4が好ましい。
A/(B+C)の値が0.9より小さいと、焼成工程でシリカに転化したときの収縮率の充分な低減効果が得られない。またこの値が1.5より大きくても同様である。
(A+B)/Cの値が0.9より大きいと収縮率が小さくなる理由は、ユニットN−3がシリカへ転化するときに、窒素1分子が、酸素1.5分子と置き換わることにより、ユニットが占める体積が増加することに起因すると、我々は考察している。
A/(B+C)の値が1.5より大きいと、収縮率の低減が得られない理由は、ユニットN−3が増加すると、無機ポリシラザンがシリカへ転化する場合に必要なアンモニア分子が少なくなり、その結果、無機ポリシラザン中のSi−N結合がSi−O結合に転化される割合が小さくなり、シリカに転化されなかったポリシラザン部分が、アウトガスとしてロスすることになり、ユニットN−3の収縮抑制の効果を打ち消すことになると、我々は考察している。
【0025】
本発明の無機ポリシラザンにおける(A+B)/Cの値は、4.2〜50であり、4.5〜20が好ましい。
(A+B)/Cの値が4.2より小さいと、焼成工程でシリカに転化したときの収縮率が大きくなる。また、この値が50より大きい無機ポリシラザンの製造は、困難である。(A+B)/Cの値が小さいということは、SiH3基が多いということになり、SiH3基は、シリカ転化時には、分解され、モノシランのアウトガスとしてロスすることになる。(A+B)/Cの値が50より大きい無機ポリシラザンの製造が困難な理由は、アンモニアとハロシランの反応時にハロシランの一部がポリマー化反応前に不均化反応を起こし、珪素原子に隣接する水素原子の数が変化する為であると、我々は考察している。
【0026】
本発明の無機ポリシラザンの分子量は、ポリスチレン換算値による重量平均分子量が2000〜20000であり、3000〜10000が好ましい。
重量平均分子量が2000より小さいと、シリカ膜形成時の乾燥工程や焼成工程において塗膜からのアウトガスが増え、シリカ膜の膜厚の低下や亀裂の発生が起こる。20000より大きいと、微細なパターンやアスペクト比の大きいパターンの埋め込み性が悪化し、良好なシリカ膜の形成が難しくなる。
【0027】
また、本発明の無機ポリシラザン中の低分子量成分が過剰に多い場合は、乾燥工程や焼成工程において塗膜からの揮発物又は昇華物が増え、シリカ膜の膜厚の低下や亀裂の発生が起こる場合があることから、本発明の無機ポリシラザン中の質量平均分子量が800以下である成分の割合は、40%以下であることが好ましく、30%以下であることが更に好ましい。
【0028】
尚、本発明において、質量平均分子量とは、テトラヒドロフラン(THF)を溶媒とし、示差屈折率検出器(RI検出器)を用いてGPC分析を行った場合のポリスチレン換算の質量平均分子量をいう。また、本発明の無機ポリシラザン中の質量平均分子量が800以下である成分の割合とは、GPC分析を行った場合の無機ポリシラザンのピーク面積比で、全ポリシラザン量に対する、ポリスチレン換算で質量平均分子量800以下のポリシラザン量の割合をいう。
【0029】
本発明の無機ポリシラザンの赤外スペクトルにおいて、Si−H結合に由来する吸収が2050〜2400cm-1に、N−H結合に由来する吸収が3300〜3450cm-1にある。従って、2050〜2400cm-1の吸光度は、珪素原子に結合する水素原子の数に対応し、3300〜3450cm-1の吸光度は、窒素原子に結合する水素原子の数に対応するので、赤外スペクトルにおける2050〜2400cm-1の範囲で最大の吸光度に対する3300〜3450cm-1の範囲で最大の吸光度の比は、窒素原子に結合する水素原子数/珪素原子に結合する水素原子数の指標となる。本発明では、以降この比をNH/SiH吸光度比という。
【0030】
本発明の無機ポリシラザンにおいて、NH/SiH吸光度比が0.01よりも小さいと、本発明の無機ポリシラザンの保存安定性が不良となる場合があり、0.20よりも大きいと、焼成によるシリカ転化時の収縮が大きくなる場合があることから、NH/SiH吸光度比は、0.01〜0.20であることが好ましく、0.10〜0.20であることがより好ましい。
【0031】
本発明における無機ポリシラザンの赤外スペクトルは、透過法及び反射法の何れで測定してもよい。透過法により測定する場合には、2050〜2400cm-1及び3300〜3450cm-1に、実質的に赤外スペクトルの妨害吸収のない試験片に、無機ポリシラザンを塗布した後、赤外スペクトルを測定することにより得ることができる。反射法により測定する場合も透過法と同様の試験片で測定が可能であるが、透過法に比べてS/N比が劣る場合がある。簡便で再現性が良好な方法は、例えば、両面研磨したシリコンウェハーを基体とし、スピンコーターで塗布して乾燥させた無機ポリシラザンを透過により、測定する方法である。
【0032】
上記基体上に形成される無機ポリシラザンの膜厚は、300〜1000nmの場合に精度よくNH/SiH吸光度比が得られる。赤外スペクトルの測定には、測定後のデータ処理が容易であることから、フーリエ変換型赤外分光計(FT−IR)を用いることが好ましい。
【0033】
本発明におけるNH/SiH吸光度比は、無機ポリシラザンの赤外スペクトルのスペクトルチャートから頂点強度法により得られた値である。例えば、図1において、2050cm-1、2400cm-1、3300cm-1及び3450cm-1における、吸光度曲線上の点をそれぞれ点A、点B、点E及び点Fとし、2050〜2400cm-1の範囲及び3300〜3450cm-1の範囲で吸光度が最大となる波数の、吸光度曲線上の点をそれぞれ点C及び点Gとし、点Cから基準線(吸光度0となる線、ブランク)への垂線と線ABとの交点を点D、点Gから基準線への垂線と線EFとの交点を点Hとするとき、NH/SiH吸光度比は、線分CDに対する線分GHの比に相当する。即ち、本発明のNH/SiH吸光度比は、無機ポリシラザンの赤外スペクトルのスペクトルチャートにおいて、2050cm-1の吸光度の点と2400cm-1の吸光度の点を結ぶ線をベースラインとした2050〜2400cm-1の吸光度最大値に対する、3300cm-1の吸光度の点と3450cm-1の吸光度の点を結ぶ線をベースライン3300〜3450cm-1の吸光度最大値の比である。
尚、通常、無機ポリシラザンは、2050〜2400cm-1の範囲で吸光度が最大となるのは2166cm-1付近であり、3300〜3450cm-1の範囲で吸光度が最大となるのは3377cm-1付近である。
【0034】
本発明の無機ポリシラザンは、波長633nmにおける屈折率が、1.550よりも小さい場合には、焼成によるシリカ転化時の収縮が大きくなる場合があり、1.650よりも大きい場合には、本発明のシリカ膜形成用塗布液の保存安定性が不良となる場合があるので、波長633nmにおける屈折率は、1.550〜1.650であることが好ましく、1.560〜1.640であることが更に好ましく、1.570〜1.630であることが最も好ましい。
【0035】
上記の屈折率の測定方法は、例えば、無機ポリシラザン又は無機ポリシラザンを溶解又は分散せしめた組成物を基体上に、スピンコート法、ディップコート法、ナイフコート法、ロールコート法等の方法により塗布し、乾燥して無機ポリシラザン膜を形成し測定すればよい。乾燥は、無機ポリシラザン膜の膜厚によって異なるが、500〜1000nmの場合には、150℃で1分以上、好ましくは150℃で3分程度加熱することが好ましい。ケイ素含量に対する窒素含量の比が同一である無機ポリシラザンの場合、屈折率の高い無機ポリシラザンの方が、水素含量が少なく、分子中に多数の環構造を有しており、これが、シリカ膜形成用塗布液の保存安定性や水蒸気中における焼成工程での収縮に影響を与えているものと考えられる。
【0036】
本発明の無機ポリシラザンの製造方法は、特に制限されず周知の無機ポリシラザンの製造方法を適用又は応用して製造すればよい。例えば、ハロシラン化合物とアンモニアとを直接反応させてもよく、ハロシラン化合物に塩基等の付加物を付加させた付加体を形成し、その付加体とアンモニアとを反応させてもよい。このような付加体を経由する無機ポリシラザンの製造方法は、例えば、特開昭60−145903号公報、特開昭61−174108号公報等に開示されている。
本発明の無機ポリシラザンの製造方法としては、反応が制御できる点から、ハロシラン化合物と塩基とを反応させて付加体を形成した後、該付加体とアンモニアとを反応させる方法が好ましい。
【0037】
ハロシラン化合物と塩基とを反応させて付加体を形成した後、該付加体とアンモニアとを反応させる無機ポリシラザンの製造方法において、当該付加体とアンモニアとの反応は、通常、−50〜20℃、好ましくは−10〜15℃の温度で行う。
【0038】
本発明の無機ポリシラザンの原料に使用するハロシラン化合物としては、ジクロロシラン、ジブロモシラン、クロロブロモシラン等のジハロシラン化合物;トリクロロシラン、トリブロモシラン、ジクロロブロモシラン、クロロジブロモシラン等のトリハロシラン化合物、テトラクロロシラン、テトラブロモシランが挙げられるが、ハロシランとしては、クロロシラン類が安価であるので好ましい。ハロシラン化合物は1種のみを使用してもよいし、2種以上を組み合わせて使用してもよい。ジハロシラン化合物を使用した無機ポリシラザンは成膜性に優れ、トリハロシラン化合物を使用した無機ポリシラザンは焼結時の収縮が少ないという利点があり、本発明の無機ポリシラザンを製造する場合には、ジハロシラン化合物、トリハロシラン化合物、又はジハロシラン化合物とトリハロシラン化合物を混合して使用することが好ましい。
【0039】
ジハロシラン化合物とトリハロシラン化合物を混合して使用する場合、ユニットS−2の数を制御する点から、その割合が、ジハロシラン化合物1モルに対してトリハロシラン化合物が、0.01〜2モルであることが好ましく、0.03〜1モルであることが更に好ましく、0.05〜0.5モルであることが最も好ましい。
【0040】
また、付加体を形成するための付加物である塩基は、ハロシラン化合物との付加体を形成する反応以外は不活性である塩基がよい。このような塩基としては、例えば、トリメチルアミン、トリエチルアミン、トリブチルアミン、ジメチルアニリン等の3級アミン類;ピリジン、ピコリン等のピリジン類が挙げられ、工業的な入手の容易さと取扱いの容易さの点から、ピリジン及びピコリンが好ましく、ピリジンが更に好ましい。使用する塩基の量は、ハロシラン化合物のハロゲン原子に対して、1倍モル以上であればよいが、付加体の形成が不十分とならないよう、1.1倍モル以上であることが好ましい。
【0041】
本発明の無機ポリシラザンの製造方法において、上記付加体が形成されると、反応系の流動性が低下するので、付加体の形成反応は有機溶剤中で行うことが好ましい。当該溶剤は、無機ポリシラザンと反応しない有機溶剤を使用することができる。例えば、ペンタン、ヘキサン、ヘプタン、オクタン、2,2,4−トリメチルペンタン(イソオクタンともいう)、イソノナン、2,2,4,6,6−ペンタメチルヘプタン(イソドデカンともいう)等の飽和鎖状炭化水素化合物;シクロペンタン、シクロヘキサン、メチルシクロヘキサン、エチルシクロヘキサン、デカリン等の飽和環状炭化水素化合物;ベンゼン、トルエン、キシレン、エチルベンゼン、クメン、プソイドクメン、テトラリン等の芳香族炭化水素化合物;ジエチルエーテル、ジプロピルエーテル、ジイソプロピルエーテル、ジブチルエーテル、ジイソブチルエーテル、t−ブチルメチルエーテル、テトラヒドロフラン、ジオキサン、1,2−ジメトキシエタン等のエーテル化合物等が挙げられる。
【0042】
また、有機溶剤の代わりの反応溶媒として、付加物である塩基を過剰量用いて、過剰量の塩基を溶媒としてもよい。特に好ましいのは、ピリジンを付加物として、これを形成反応が終了しても流動性を保てる程度に過剰量使用し、他の有機溶剤を使用しないことである。この場合、ピリジンの使用量は、ハロシラン化合物に対して3〜30倍モルであることが好ましく、4〜25倍モルであることが更に好ましく、5〜20倍モルであることが好ましい。また、付加体形成による流動性の低下を防ぐため、有機溶剤、付加物、有機溶剤と付加物を含有する混合溶媒に、ハロシラン化合物とアンモニアとを分割して仕込んでもよく、又は連続的に同時に仕込んでもよい。
【0043】
付加体を経由する製造方法においては、アンモニアの使用量は、化学量論上、反応に使用するハロシラン化合物のハロゲン原子に対して等モル以上(1倍モル以上)であればよいが、反応を完結させるのに十分であり経済性を考慮すると、アンモニアの使用量は、反応に使用するハロシラン化合物のハロゲン原子に対して、1.0〜3.0倍モルであることが好ましく、1.1〜2.5倍モルであることが更に好ましく、1.2〜2.0倍モルであることが最も好ましい。
【0044】
アンモニアとの反応後は、必要に応じて過剰のアンモニアを除去し、生成したハロゲン化アンモニウムを濾過等により除去する。その後、必要に応じて公知の方法により、所望の有機溶剤に溶剤置換等を行えばよい。
【0045】
また、本発明の無機ポリシラザンは、生成した塩の除去前、又は除去後に、無機ポリシラザン分子中のSiH基とNH基とを反応させてSi−N結合を生成させることにより、分子内反応による環状化、分子間反応による高分子量化等を行ってもよく、これにより、SiH3基の減少、質量平均分子量の増加、質量平均分子量が800以下である成分の減少、NH/SiH吸光度比の増加、屈折率の増加等の調整を図ってもよい。無機ポリシラザンのSiH基とNH基を反応させてSi−N結合を生成させる方法としては、例えば、ピリジン、ピコリン等の塩基性溶媒中で加熱する方法(例えば、特開平1−138108号公報を参照)、アルカリ金属水素化物、アルカリ金属アルコキシド、無水アルカリ金属水酸化物等のアルカリ金属含有塩基性触媒による方法(例えば、特開昭60−226890号公報を参照)、テトラメチルアンモニウムヒドロキシド等の4級アンモニウム化合物を触媒とする方法(例えば、特開平5−170914号公報を参照)、硝酸アンモニウム、酢酸アンモニウム等の酸触媒を使用する方法(例えば、特開表2003−514822号公報を参照)等が挙げられ、反応に使用した付加物又は当該付加物を含有する溶媒中で加熱する方法が好ましい。
【0046】
付加体を経由する方法で無機ポリシラザンを製造する場合には、付加体(例えばジクロロシランとピリジン)とアンモニアとが反応して付加物(例えばピリジン)が遊離することから、遊離した付加物を塩基性溶媒として使用することが可能である。従って、付加体を経由して無機ポリシラザンを製造した後、遊離した付加物を溶媒として加熱することにより無機ポリシラザンのSiH基とNH基を反応させてSi−N結合を生成させることは、原料の有効利用及び製造工程の簡略化の点から好ましい。
【0047】
本発明のシリカ膜形成用塗布剤は、上記の本発明の無機ポリシラザンと、有機溶剤を必須成分として含有する組成物であり、基体に塗布しやすい濃度に調製される。
【0048】
本発明のシリカ膜形成用塗布剤に使用される有機溶剤は、無機ポリシラザンと反応することで塗布性を損ねる程の変質や反応をきたすものでなければ、特に限定されない。水酸基、アルデヒド基、ケトン基、カルボキシル基、エステル基等は、無機ポリシラザンとの高い反応性を有することから、これらの基を有しないものが好ましい。本発明のシリカ膜形成用塗布液の好ましい有機溶剤としては、例えば、ペンタン、ヘキサン、ヘプタン、オクタン、2,2,4−トリメチルペンタン(イソオクタンともいう)、イソノナン、2,2,4,6,6−ペンタメチルヘプタン(イソドデカンともいう)等の飽和鎖状炭化水素化合物;シクロペンタン、シクロヘキサン、メチルシクロヘキサン、デカリン等の飽和環状炭化水素化合物;ベンゼン、トルエン、キシレン、エチルベンゼン、クメン、プソイドクメン、テトラリン等の芳香族炭化水素化合物;ジエチルエーテル、ジプロピルエーテル、ジイソプロピルエーテル、ジブチルエーテル、ジイソブチルエーテル、t−ブチルメチルエーテル、テトラヒドロフラン、ジオキサン、1,2−ジメトキシエタン等のエーテル化合物等が挙げられ、中でも、塗布性が良好であることから、キシレン、ジブチルエーテルが好ましく、保存安定性が良好であることからジブチルエーテルが更に好ましい。有機溶剤は、1種類のみでもよいが、蒸発速度の調整等の目的で2種以上を組み合わせて用いてもよい。
【0049】
エーテル化合物は、その原料、製造工程の副生成物、保存中の劣化生成物として、アルコール化合物、アルデヒド化合物、ケトン化合物、カルボン酸化合物、エステル化合物等が含まれることがある。本発明のシリカ膜形成用塗布液の有機溶剤に、これらの化合物が含まれる場合には焼成工程での収縮が大きくなる場合があることから、無機ポリシラザンと混合する以前に、これらのアルコール化合物、アルデヒド化合物、ケトン化合物、カルボン酸化合物及びエステル化合物の含量の合計を、ジブチルエーテルに対して0.1質量%以下にしておくことが好ましく、0.05質量%以下であることが更に好ましく、0.01質量%以下であることが最も好ましい。
【0050】
本発明のシリカ膜形成用塗布液中の無機ポリシラザンの含量が、あまりに低い場合には、シリカ膜の成膜性が不十分となり、あまりに高い場合には本発明のシリカ膜形成用塗布液の保存安定性が不十分となりゲル物が発生する場合があることから、無機ポリシラザンの含量は、1〜40質量%が好ましく、3〜35質量%が更に好ましく、5〜30質量%が最も好ましい。
【0051】
本発明のシリカ膜形成用塗布液は、主として、該塗布液を基体(対象材料)上に塗布し、該塗布液と、酸化剤とを反応させて、形成されるシリカ膜として、従来、無機ポリシラザンが使用されてきた用途、例えば、半導体装置の絶縁膜、フラットパネルディスプレイの保護膜、光学関連製品の反射防止膜等に使用でき、特に半導体装置の絶縁膜に好適に使用できる。
【0052】
例えば、半導体装置の絶縁膜を形成する場合には、本発明のシリカ形成用塗布液を対象材料(基体)上に塗布し、塗膜を形成する塗布工程、塗膜から有機溶媒を除去する乾燥工程、水蒸気中において焼成しシリカ膜を形成する焼成工程を含む製造方法が好ましい。
本発明のシリカ膜形成用塗布液を対象材料に塗布する場合には、特に限定されず、スプレー法、スピンコート法、ディップコート法、ロールコート法、フローコート法、スクリーン印刷法、転写印刷法等のいずれの塗布方法でもよいが、膜厚が薄く均一な塗膜が形成できることからスピンコート法が好ましい。
【0053】
乾燥工程の乾燥温度及び時間は、使用する有機溶媒及び塗膜の膜厚により異なるが、80〜200℃、好ましくは120〜170℃において、1〜30分、好ましくは2〜10分加熱することが好ましい。乾燥雰囲気は、酸素中、空気中、不活性ガス中の何れでもよい。また、焼成工程は、相対湿度20〜100%の水蒸気雰囲気下に200〜1200℃の温度が好適な範囲である。焼成温度が低い場合には、反応が十分進行しない場合があるとともに、シラノール基の残存による絶縁性の低下の懸念があり、焼成温度が高いと製造コストの問題があることから、水蒸気雰囲気下の焼成の温度は、300〜1000℃が好ましく、700〜900℃が更に好ましい。焼成する場合は、700℃以上の温度により1段階で焼成してもよいし、200〜500℃、好ましくは300〜450℃で30〜60分焼成した後に、450〜1200℃、好ましくは600〜1000℃、更に好ましくは700〜900℃で焼成する2段階の焼成でもよい。シリカ膜の収縮が少なく、亀裂が生じにくいことから2段階の焼成が好ましい。このほか、200〜500℃、好ましくは350〜450℃で30〜60分焼成した後に、20〜80℃の蒸留水に浸漬させる低温焼成法(例えば、特開平7−223867号公報を参照)でもよいが、低温焼成法ではシラノール基の残存による絶縁性の低下が起こることから、低温焼成後に大気中で700〜900℃で5〜60分程度加熱することが好ましい。
【実施例】
【0054】
以下、実施例により本発明を具体的に説明するが、これらは本発明の範囲を限定するものではない。尚、実施例中の「部」や「%」は質量によるものである。また、溶媒に用いたジブチルエーテルは純度が99.99%であり、アルコール化合物、アルデヒド化合物、ケトン化合物、カルボン酸化合物及びエステル化合物の合計の含量が0.01%以下であるものを用いた。
【0055】
[実施例1]
攪拌機、温度計及び導入管を備えた3000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン2310g(29.2モル)を仕込み、撹拌しながら、トリクロロシラン48.6g(0.36モル)とジクロロシラン82.6g(0.82モル)を反応温度0〜5℃となるように冷却しながら、それぞれ1時間かけて滴下し、ピリジン付加体を生成させた。アンモニア78.9g(4.64モル)を、反応温度が10℃を超えないように冷却しながら3時間かけて導入管からフィードし、窒素ガスを吹き込みながら、更に10℃で1.5時間撹拌を行い、反応を完結させた。この反応液を10℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾別し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに溶媒交換をした。得られた溶液を、120℃で6時間加熱した後、濾過径0.1μmのPTFE製カートリッジフィルターで濾過を行い、無機ポリシラザン含量が11.3%であるシリカ膜形成用塗布液No.1を得た。
【0056】
[実施例2]
攪拌機、温度計及び導入管を備えた3000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン2310g(29.2モル)を仕込み、撹拌しながら、トリクロロシラン50.4g(0.37モル)とジクロロシラン82.9g(0.82モル)を、反応温度が−10〜0℃となるように冷却しながら、それぞれ1時間かけて滴下し、ピリジン付加体を生成させた。アンモニア78.9g(4.61モル)を、反応温度が5℃を超えないように冷却しながら3時間かけて導入管からフィードし、窒素ガスを吹き込みながら、更に10℃で1.5時間撹拌を行い、反応を完結させた。この反応液を10℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾別し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに溶媒交換をした。得られた溶液を、120℃で6時間加熱した後、濾過径0.1μmのPTFE製カートリッジフィルターで濾過を行い、無機ポリシラザン含量が18.7%であるシリカ膜形成用塗布液No.2を得た。
【0057】
[実施例3]
攪拌機、温度計及び導入管を備えた3000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン2411g(30.5モル)を仕込み、撹拌しながら、トリクロロシラン69.8g(0.52モル)とジクロロシラン51.3g(0.51モル)を、反応温度が−10〜0℃となるように冷却しながら、それぞれ1時間かけて滴下し、ピリジン付加体を生成させた。アンモニア74.4g(4.35モル)を、反応温度−10〜0℃で3時間かけて導入管からフィードし、窒素ガスを吹き込みながら、更に10℃で1.5時間撹拌を行い、反応を完結させた。この反応液を10℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾別し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに溶媒交換をした。得られた溶液を、120℃で6時間加熱した後、濾過径0.1μmのPTFE製カートリッジフィルターで濾過を行い、無機ポリシラザン含量が9.64%であるシリカ膜形成用塗布液No.3を得た。
【0058】
[比較例1]
攪拌機、温度計及び導入管を備えた3000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン1646g(20.8モル)を仕込み、撹拌しながら、ジクロロシラン310g(3.1モル)を、反応温度0〜5℃で1時間かけて導入管からフィードし、ジクロロシランのピリジンアダクツを生成させた。アンモニア180g(10.6モル)を、反応温度0〜5℃で1時間かけて導入管からフィードし、更に10℃で1.5時間撹拌を行い、反応を完結させた。この反応液を10℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾過し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに溶媒交換をした。得られた溶液を、窒素雰囲気下、濾過径0.1μmのPTFE製カートリッジフィルターにて濾過を行い、無機ポリシラザン含量が19.0%である比較用塗布液1を得た。
【0059】
[比較例2]
攪拌機、温度計及び導入管を備えた3000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン2248g(28.4モル)を仕込み、撹拌しながら、ジクロロシラン191.0g(1.89モル)とアンモニア113.0g(6.65モル)を、反応温度が0〜5℃となるように冷却しながら、それぞれ3時間かけてフィードし、窒素ガスを吹き込みながら、更に10℃で1.5時間撹拌を行い、反応を完結させた。この反応液を10℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾別し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに溶媒交換をした。得られた溶液を、120℃で6時間加熱した後、濾過径0.1μmのPTFE製カートリッジフィルターで濾過を行い、無機ポリシラザン含量が19.2%である比較用塗布液2を得た。
【0060】
[比較例3]
攪拌機、温度計及び導入管を備えた3000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン2044g(25.8モル)を仕込み、撹拌しながら、ジクロロシラン174.0g(1.72モル)とアンモニア103.0g(6.06モル)を、反応温度が0〜5℃となるように冷却しながら、それぞれ3時間かけてフィードし、窒素ガスを吹き込みながら、更に10℃で1.5時間撹拌を行い、反応を完結させた。この反応液を10℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾別し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに溶媒交換をした。得られた溶液を、120℃で6時間加熱した後、濾過径0.1μmのPTFE製カートリッジフィルターで濾過を行い、無機ポリシラザン含量が19.3%である比較用塗布液3を得た。
【0061】
[比較例4]
攪拌機、温度計及び導入管を備えた3000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン2303g(29.1モル)を仕込み、撹拌しながら、ジクロロシラン280.0g(2.77モル)とアンモニア165.0g(9.71モル)を、反応温度が−10〜0℃となるように冷却しながら、それぞれ4時間かけてフィードし、窒素ガスを吹き込みながら、更に0℃で1.5時間撹拌を行い、反応を完結させた。この反応液を10℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾別し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに溶媒交換をした。得られた溶液を、120℃で6時間加熱した後、濾過径0.1μmのPTFE製カートリッジフィルターで濾過を行い、無機ポリシラザン含量が19.0%である比較用塗布液4を得た。
【0062】
[比較例5]
攪拌機、温度計及び導入管を備えた3000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン2044g(25.8モル)を仕込み、撹拌しながら、ジクロロシラン325.7g(3.22モル)とアンモニア192.1g(11.3モル)を、反応温度が−10〜0℃となるように冷却しながら、それぞれ2時間かけてフィードし、窒素ガスを吹き込みながら、更に0℃で1.5時間撹拌を行い、反応を完結させた。この反応液を10℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾別し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに溶媒交換をした。得られた溶液を、120℃で6時間加熱した後、濾過径0.1μmのPTFE製カートリッジフィルターで濾過を行い、無機ポリシラザン含量が19.2%である比較用塗布液5を得た。
【0063】
[比較例6]
攪拌機、温度計及び導入管を備えた3000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン2044g(25.8モル)を仕込み、撹拌しながら、ジクロロシラン260.6g(2.58モル)とアンモニア131.6g(7.74モル)を、反応温度が−10〜0℃となるように冷却しながら、それぞれ1.5時間かけてフィードし、窒素ガスを吹き込みながら、更に0℃で1.5時間撹拌を行い、反応を完結させた。この反応液を10℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾別し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに溶媒交換をした。得られた溶液を、120℃で6時間加熱した後、濾過径0.1μmのPTFE製カートリッジフィルターで濾過を行い、無機ポリシラザン含量が20.3%である比較用塗布液6を得た。
【0064】
[比較例7]
攪拌機、温度計及び導入管を備えた5000mlのガラス製反応容器に、窒素雰囲気中、乾燥ピリジン4300g(54.4モル)を仕込み、撹拌しながら、ジクロロシラン545g(5.4モル)を、反応温度−40〜−30℃で1時間かけて導入管からフィードし、ジクロロシランのピリジンアダクツを生成させた。アンモニア325g(19.1モル)を、反応温度−40〜−30℃で1時間かけて導入管からフィードし、更に−20〜−15℃で2時間撹拌を行い、反応を完結させた。この反応液を25℃に加熱した後、窒素雰囲気中、生成した塩化アンモニウムを濾過し、過剰のアンモニアを減圧除去してから、溶媒をピリジンからジブチルエーテルに常法により交換し、更にアルゴンガス雰囲気中で、濾過径0.1μmのPTFE製カートリッジフィルターにて濾過を行い、無機ポリシラザン含量が19.0%である比較用塗布液7を得た。
【0065】
[比較例8]
比較例7において、アンモニアの反応温度を−40〜−30℃から−15〜−12℃に変更し、その後−15〜−12℃で2時間撹拌した以外は、比較例7と同様の操作を行い、無機ポリシラザン含量が19.1%である比較用塗布液8を得た。
【0066】
[比較例9]
比較例7において、ジクロロシラン545g(5.4モル)の代わりにジクロロシラン444g(4.4モル)とトリクロロシラン13.6g(1.0モル)との混合物を使用し、アンモニアを325g(19.1モル)から340g(20.0モル)に増やした以外は、比較例7と同様の操作を行い、無機ポリシラザン含量が19.2%である比較用塗布液9を得た。
【0067】
<分析:1H―NMR分析>
実施例1〜3で得たシリカ膜形成用塗布液No.1〜3及び比較例1〜9で得た比較用塗布液1〜9について、1H―NMRを測定した。シリカ膜形成用塗布液No.1〜No.4のチャートを図2〜図4に示す。1H―NMRスペクトルにおいて、4.75ppm以上で5.4ppm未満の範囲のピーク面積をAとし、4.5ppm以上で4.75ppm未満の範囲のピーク面積をB、4.2ppm以上で4.5ppm未満の範囲のピーク面積をCとして、A/(B+C)の値、(A+B)/Cの値を算出した。結果を〔表1〕に示す。
【0068】
<分析:GPC>
実施例1〜3で得たシリカ膜形成用塗布液No.1〜3及び比較例1〜9で得た比較用塗布液1〜9について、GPCの結果から、無機ポリシラザンの質量平均分子量、及び質量平均分子量800以下の成分の含量をそれぞれ算出した。結果を〔表1〕に示す。カラムは東ソー(株)製、スーパーマルチポアHZ−Mを使用した。
【0069】
<無機ポリシラザン塗膜の分析:膜厚、IR分析>
実施例1〜3で得たシリカ膜形成用塗布液No.1〜3及び比較例1〜9で得た比較用塗布液1〜9を、両面を研磨した厚さ4インチのシリコンウェハーに、乾燥後の無機シラザンの膜厚が580〜620nmとなるようにスピンコート法により塗布してから150℃で3分間乾燥して、無機ポリシラザンの塗膜を有するシリコンウェハーを調製し、塗膜の膜厚、FT−IRを測定した。なお、FT−IR測定では、両面を研磨したシリコンウェハーをリファレンスとした。また、膜厚は、Filmetrics社製、F−20を用いて測定した。膜厚及びFT−IRの結果から算出したNH/SiH吸光度比を〔表1〕に示す。
【0070】
【表1】
【0071】
[実施例4及び比較例10]
上記の無機ポリシラザンの塗膜の分析に用いたシリコンウェハーを用いて、1段目の焼成として、相対湿度90%で温度300℃のオーブンに30分、2段目の焼成として、相対湿度10%で温度900℃のオーブンで30分、焼成することによりシリカ絶縁膜を形成させ、シリカ膜の膜厚を測定した。乾燥後の無機シラザンの膜厚に対するシリカ絶縁膜の膜厚を硬化収縮率(%)とした。結果を〔表2〕に示す。
【0072】
【表2】
【0073】
上記〔表1〕及び〔表2〕の結果から、A/(B+C)の値、(A+B)/Cの値及び質量平均分子量が所定の範囲内にある、本願発明の無機ポリシラザンを含有するシリカ膜形成用塗布液は、A/(B+C)の値、(A+B)/Cの値及び質量平均分子量が所定の範囲内にない無機ポリシラザンを含有する比較用塗布液に比して、硬化収縮率が小さく、シリカ膜の亀裂や半導体基板との剥離が発生しにくいことが明らかである。
【特許請求の範囲】
【請求項1】
1H―NMRスペクトルにおいて、4.75ppm以上で5.4ppm未満の範囲のピーク面積をAとし、4.5ppm以上で4.75ppm未満の範囲のピーク面積をBとし、4.2ppm以上で4.5ppm未満の範囲のピーク面積をCとしたとき、A/(B+C)の値が0.9〜1.5であり、(A+B)/Cの値が4.2〜50であり、ポリスチレン換算値による質量平均分子量が2000〜20000である、無機ポリシラザン。
【請求項2】
赤外スペクトルにおいて、2050〜2400cm-1の範囲で最大の吸光度に対する3300〜3450cm-1の範囲で最大の吸光度の比が、0.01〜0.20である、請求項1に記載の無機ポリシラザン。
【請求項3】
ジハロシラン化合物、トリハロシラン化合物、又はこれらの混合物と塩基とを反応させて付加体を形成した後、該付加体とアンモニアとを反応させて得られる、請求項1又は2に記載の無機ポリシラザン。
【請求項4】
請求項1〜3の何れか1項に記載の無機ポリシラザンと有機溶剤とを必須成分として含有してなるシリカ膜形成用塗布液。
【請求項5】
請求項4に記載のシリカ膜形成用塗布液を基体上に塗布し、該塗布液と酸化剤とを反応させて、シリカ膜を形成することを特徴とするシリカ膜の形成方法。
【請求項1】
1H―NMRスペクトルにおいて、4.75ppm以上で5.4ppm未満の範囲のピーク面積をAとし、4.5ppm以上で4.75ppm未満の範囲のピーク面積をBとし、4.2ppm以上で4.5ppm未満の範囲のピーク面積をCとしたとき、A/(B+C)の値が0.9〜1.5であり、(A+B)/Cの値が4.2〜50であり、ポリスチレン換算値による質量平均分子量が2000〜20000である、無機ポリシラザン。
【請求項2】
赤外スペクトルにおいて、2050〜2400cm-1の範囲で最大の吸光度に対する3300〜3450cm-1の範囲で最大の吸光度の比が、0.01〜0.20である、請求項1に記載の無機ポリシラザン。
【請求項3】
ジハロシラン化合物、トリハロシラン化合物、又はこれらの混合物と塩基とを反応させて付加体を形成した後、該付加体とアンモニアとを反応させて得られる、請求項1又は2に記載の無機ポリシラザン。
【請求項4】
請求項1〜3の何れか1項に記載の無機ポリシラザンと有機溶剤とを必須成分として含有してなるシリカ膜形成用塗布液。
【請求項5】
請求項4に記載のシリカ膜形成用塗布液を基体上に塗布し、該塗布液と酸化剤とを反応させて、シリカ膜を形成することを特徴とするシリカ膜の形成方法。
【図1】

【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公開番号】特開2013−1721(P2013−1721A)
【公開日】平成25年1月7日(2013.1.7)
【国際特許分類】
【出願番号】特願2011−131146(P2011−131146)
【出願日】平成23年6月13日(2011.6.13)
【出願人】(000000387)株式会社ADEKA (987)
【Fターム(参考)】
【公開日】平成25年1月7日(2013.1.7)
【国際特許分類】
【出願日】平成23年6月13日(2011.6.13)
【出願人】(000000387)株式会社ADEKA (987)
【Fターム(参考)】
[ Back to top ]