
高発現ベクター、バチルス属細菌、およびペプチド又は蛋白質の製造方法
【目的】 バチルス属細菌内に高発現ベクターを安定に保持させ、かつペプチド又は蛋白質を菌体外に多量に分泌生産させる。
【構成】 高発現ベクターは、ベクタープラスミドpUB110のSphI切断部位からBamHI切断部位までの塩基配列を含む。また、BamHI切断部位またはその近傍には、SphI切断部位からBamHI切断部位への方向、すなわち、従来とは逆方向であるカナマイシン耐性遺伝子の方向に、SD配列を有するペプチド又は蛋白質をコードする遺伝子を連結する。前記高発現ベクターによりバチルス属細菌を形質転換し、形質転換株を培養すると、多量のペプチド又は蛋白質が得られる。
【構成】 高発現ベクターは、ベクタープラスミドpUB110のSphI切断部位からBamHI切断部位までの塩基配列を含む。また、BamHI切断部位またはその近傍には、SphI切断部位からBamHI切断部位への方向、すなわち、従来とは逆方向であるカナマイシン耐性遺伝子の方向に、SD配列を有するペプチド又は蛋白質をコードする遺伝子を連結する。前記高発現ベクターによりバチルス属細菌を形質転換し、形質転換株を培養すると、多量のペプチド又は蛋白質が得られる。
【発明の詳細な説明】
【0001】
【産業上の利用分野】本発明は、枯草菌を代表とするバチルス属細菌で利用可能な新規な高発現ベクター、このベクターを保有するバチルス属細菌、およびこのバチルス属細菌によるペプチド又は蛋白質の製造方法に関する。
【0002】
【従来の技術と発明が解決しようとする課題】枯草菌(Bacillus subtilis)を代表とするバチルス(Bacillus)属細菌は、古くから種々の菌体外酵素の生産菌として、工業的に利用されている。例えば、枯草菌の仲間は、日本において古くから納豆の製造に使用されており、大腸菌などで問題となるエンドトキシンを生産しないこと、人体に寄生しないことなどの理由から、極めて安全性の高い微生物である。このように、枯草菌は、ペプチド又は蛋白質の工業的な生産菌として、優れた性質を有し、その有用性が注目されている。例えば、特開昭56−137986号公報には、前記菌体外酵素の遺伝子のプロモーター、SD配列(シャイン−ダルガーノ(Shine-Dalgarno)配列)、シグナル遺伝子をコードするDNAをクローニングし、その下流に目的とするペプチド又は蛋白質をコードする遺伝子を連結し、これを枯草菌に導入し、目的とするペプチド又は蛋白質を菌体外に分泌生産させている。
【0003】しかしながら、枯草菌におけるペプチド又は蛋白質の生産は、枯草菌自体が産生するα−アミラーゼやプロテアーゼなどが主である。すなわち、枯草菌が高い分泌生産能を有するにも拘らず、他の微生物由来の酵素及び比較的分子量の小さな哺乳類の生理活性ペプチドなどを生産させると、菌体自体が菌体外に多量に分泌する各種のプロテアーゼにより生産物が分解され、高い収率で有用生産物を回収できない。
【0004】そこで、宿主枯草菌のプロテアーゼ活性を低下させるための研究がなされている。例えば、プロテアーゼ活性の低い枯草菌として、枯草菌の主たる菌体外プロテアーゼである中性プロテアーゼ及びアルカリプロテアーゼの両者を欠失したバチルス・サブチリス(Bacillus subtilis)104HL株[バイオケミカル・アンド・バイオフィジカル・リサーチ・コミュニケーションズ(Biochem. Biophys.Res. Commun.), 128; 601-606, 1985]が知られている。また、本出願人は、特開昭64−37284号公報において、バチルス・サブチリス104HL株に、pap 遺伝子を導入したバチルス・サブチリスDY−16株(微工研菌寄第9488号)を提案した。
【0005】一方、枯草菌ベクターの開発も、宿主の開発と同様に古くから行なわれている。例えば、枯草菌自体や、バチルス・アミロリクエファシエンス、バチルス・リケニホルミスなどの類縁菌のα−アミラーゼ、または中性及びアルカリプロテアーゼの遺伝子のプロモーター、リボソーム結合領域(SD配列)、シグナル配列を利用した分泌ベクターが発表されている。これらの分泌ベクターにより、それぞれ本来のα−アミラーゼやプロテアーゼの分泌だけでなく、例えばプロテインAなどの他の有用酵素、およびインターフェロン、ヒト成長ホルモンなどの生理活性ペプチドを生産できる。
【0006】しかしながら、枯草菌が分泌生産という有用性を有するにも拘らず、大腸菌に比べて、遺伝子組換えの宿主としての利用が遅れている。このことは、遺伝子産物の生産量が少ないことに起因する。すなわち、大腸菌系では、多くの強いプロモーター系と、極めて正確な制御系とが確立され、これらが多コピープラスミド上で安定に機能している。これに対して、枯草菌の系においては、多くの試みにも拘らず、大腸菌のように確立された系は、存在しない。
【0007】
【発明が解決しようとする課題】従って、本発明の目的は、バチルス属細菌内に安定に保持され、かつペプチド又は蛋白質を菌体外に多量に分泌生産させることができる高発現ベクターを提供することにある。
【0008】本発明の他の目的は、前記高発現ベクターを保有し、ペプチド又は蛋白質を菌体外に多量に分泌生産するバチルス属細菌を提供することにある。
【0009】本発明のさらに他の目的は、ペプチド又は蛋白質を効率よく多量に得ることができるペプチド又は蛋白質の製造方法を提供することにある。
【0010】
【発明の構成】本発明者らは、高い分泌能を有するバチルス属細菌に着目し、その分泌ベクターの開発を鋭意研究した結果、枯草菌において一般に使用されているベクタープラスミドpUB110のクローニングサイトBamHI切断部位またはその近傍に、高い翻訳能力を有する遺伝子を特定の方向に導入すると、分泌ベクターが少なくともバチルス属細菌内で安定に保持され、かつ分泌ベクターにより所望の遺伝子産物が多量に分泌発現することを見いだし、本発明を完成した。
【0011】すなわち、本発明は、少なくともpUB110のSphI切断部位からBamHI切断部位までの塩基配列を含むpUB110由来のベクターであって、BamHI切断部位またはその近傍に、SphI切断部位からBamHI切断部位への方向に、SD配列を有するペプチド又は蛋白質をコードする遺伝子が連結されている高発現ベクターを提供する。
【0012】本発明は、前記高発現ベクターを保有するバチルス属細菌を提供する。
【0013】さらに本発明は、前記バチルス属細菌を培養し、産生したペプチド又は蛋白質を回収するペプチド又は蛋白質の製造方法を提供する。
【0014】本明細書において、塩基配列における核酸の表示はIUPACにより採択されている表示法に従うものとする。
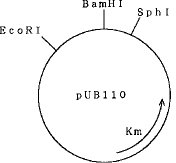
【0015】本発明の高発現ベクターは、ベクタープラスミドpUB110に由来し、pUB110のSphI切断部位からBamHI切断部位までの塩基配列を含んでいる。pUB110はその全塩基配列を有している必要はなく、少なくともSphIおよびBamHI切断部位を有していればよく、SphI、BamHIおよびEcoRI切断部位を有するpUB110由来のベクターであってもよい。なお、pUB110は黄色ブドウ状球菌[スタフィロコッカス(Staphylococcus ) ]属に由来する多コピープラスミドであり、カナマイシン耐性遺伝子を有し、その制限地図は確立されている。図1にベクタープラスミドpUB110の制限酵素地図の概要を示す。
【0016】pUB110とペプチド又は蛋白質をコードする遺伝子との連結部位は、制限酵素BamHI部位に限らず、その近傍、例えば、BamHI部位からEcoRI部位までの間などであってもよい。すなわち、EcoRI部位や、BamHI部位からEcoRI部位までの領域に存在するAvaI部位、XbaI部位に、ペプチド又は蛋白質をコードする遺伝子を連結してもよい。
【0017】クローニングサイトBamHI切断部位またはその近傍には、高い翻訳能力を有する遺伝子、すなわちSD配列を有するペプチドまたは蛋白質をコードする遺伝子が連結されている。
【0018】本発明の主たる特徴は、前記ペプチド又は蛋白質をコードする遺伝子が、従来とは逆の方向であるSphI切断部位からBamHI切断部位への方向、すなわちカナマイシン耐性遺伝子と同じ方向に導入されている点にある。このような特定の方向にペプチド又は蛋白質をコードする構造遺伝子を導入すると、宿主微生物により、ペプチド又は蛋白質を多量に分泌生産させることができる。
【0019】前記ペプチド又は蛋白質は、特に限定されず、酵素、原核細胞および真核細胞を問わず、動物性細胞由来のペプチド又は蛋白質であってもよい。ペプチド及び蛋白質には、例えば、インスリン、ガストリン、各種オピオノイドペプチド、上皮細胞成長因子、エンドセリン、バソアクティブ・インテスティナル・ポリペプチド[Vasoactive Intestinal Polypeptide (VIP)]、心房性利尿ホルモン(ANP)、サブスタンスP、カルシトニン、インシュリン様成長因子I,II、ガラニン、モチリン、バソプレシン、インターフェロン、IL−2などの各種の生理活性ペプチドまたはこれらの前駆体;α−アミラーゼ、プロテインA、ヒルジン、エグリンC、分泌性白血球由来プロテアーゼインヒビターなどの阻害剤、ヒトアルブミン、血液凝固因子、リンフォカイン、神経成長因子、肝細胞再生因子などの各種の分化誘導因子、成長因子などの蛋白質またはこれらの前駆体が含まれる。
【0020】前記SD配列は、特に制限されず、例えば、枯草菌の16S−rRNAの配列に対応して合成した下記のSD配列であってもよく、5′−CTAGAAAGGAGGTGATCTAGAATTC下記のSD配列などであってもよい。
【0021】5′−CTAGAAAGGAGGTGATAGAATTC5′−CTAGAAAGGAGGTGAGAATTC5′−CTAGAAAGGAGGTGAGAATAT5′−CTAGAAAGGAGGTGAGAATこれらのSD配列を導入すると、僅の発現の差異が認められるものの、ベクタープラスミドによる効果が本質的に損われるものではない。SD配列は、リボソーム結合部位として機能し、その下流には、開始コドンが存在する。
【0022】なお、従来、pUB110のBamHI切断部位が関与して、何かしらの転写が行なわれていることが知られていた[Bruchner, R.,et al., Gene 32, 151-160 (1984)およびCurran, J.F., and Stewart, C.J., Virology, 142, 98-111 (1985) ]。しかしながら、この知見を積極的に利用した例はない。このことは、各種のファクターが転写の強さについて及ぼす影響を正確に定量化できていなかったことに他ならない。
【0023】前記ペプチド又は蛋白質をコードする遺伝子を、SD配列と共に、ベクターDNAに前記特定の方向に連結することにより、プロモーターを導入しなくても、SD配列の強さに応じてペプチド又は蛋白質を生産できる。従って、強いSD配列であれば、プロモーターは必ずしも必要ではないが、SD配列の5′側にプロモーターを連結するのが好ましい。プロモーターとしては、例えば、枯草菌のα−アミラーゼ遺伝子のプロモーターなどが挙げられる。
【0024】構造遺伝子の下流、すなわちペプチド又は蛋白質をコードする遺伝子の3′側に、ターミネーターが連結されていなくても、ベクターによる効果が特に損われることはないが、構造遺伝子の下流には、ターミネーターが連結されているのが好ましい。ターミネーターは、特に限定されず、例えば、枯草菌のアルカリプロテアーゼ遺伝子のターミネーターや、これに対応して化学合成したターミネーターなどであってもよい。
【0025】本発明の高発現ベクターおよびこのベクターを保有するバチルス属細菌は、種々の方法で構築できる。例えば、pUB110を制限酵素BamHIで解裂し、アルカリホスファターゼで処理する。また、所望のペプチド又は蛋白質をコードする遺伝子の5′側にプロモーターおよびSD配列を、3′側にターミネーターを連結し、DNA断片を調製する。このDNA断片をpUB110のBamHI部位に導入する。なお、前記DNA断片とBamHI部位とのサイトが合わない場合には、両断片を平滑末端としたり、適当な制限酵素リンカーを用いればよい。pUB110にペプチド又は蛋白質をコードする遺伝子が導入されたキメラプラスミドにより、宿主微生物であるバチルス属細菌を形質転換する。得られた形質転換株から、カナマイシン耐性遺伝子と同じ方向に前記遺伝子が導入されたプラスミドを選択することにより、前記高発現ベクターを保有するバチルス属細菌が得られる。
【0026】しかし、この方法では、前記制限酵素による切断部位が、目的とするペプチド又は蛋白質ペプチドの遺伝子に存在しない場合には、pUB110のBamHI部位に構造遺伝子を両方向に導入し、得られたキメラプラスミドの中から、目的とするベクターを捜し出す必要がある。この方法では、目的蛋白質が枯草菌由来のα−アミラーゼであっても、カナマイシン耐性遺伝子と同じ方向に遺伝子が導入されたプラスミドを得る確率は、通常、比較的小さい。すなわち、枯草菌由来の構造遺伝子をカナマイシン耐性遺伝子と同じ方向に導入されたプラスミドは、逆方向に導入されたプラスミドに比べて、例えば、1/40程度しか得られない。さらに、枯草菌以外のペプチド又は蛋白質をコードする遺伝子を導入すると、枯草菌の生育阻害が顕著となり、目的とするプラスミドが得られにくくなる。このことを解決するためには、例えば、次の2つの方法が採用できる。
【0027】第1の方法では、pUB110をBamHIおよびEcoRIで切断し、アルカリホスファターゼで処理する。一方、所望のペプチド又は蛋白質をコードする遺伝子の5′側をBamHI部位、3′側をEcoRI部位となるように切断するか、又は合成DNAリンカーを用いてDNA断片を調製する。そして、pUB110のBamHIとEcoRI部位の間に、前記DNA断片を方向性をもって導入する。なお、この方法において、pUB110を制限酵素BamHIとXbaI、制限酵素XbaIとEcoRIなどで切断し、上記と同様にして、目的の遺伝子を方向性を考慮して導入してもよい。
【0028】第2の方法では、先ずpUB110を制限酵素SphIとEcoRIとで切断し、得られた約1キロベースペア(Kbp)の遺伝子断片を、大腸菌のベクター、例えば、プラスミドpUC19などのポリリンカーサイトEcoRI−SphI間に導入する。得られたプラスミドのpUB110に由来する遺伝子のうち、例えば、BamHI部位に、前記ペプチド又は蛋白質をコードする遺伝子を導入し、宿主大腸菌、例えばHB101などを形質転換する。得られた形質転換株からプラスミドを調製し、目的の方向に遺伝子が導入されたプラスミドを選択する。この際、pUB110上に、カナマイシン耐性遺伝子と同じ方向に構造遺伝子が導入されたプラスミドが多量に得られる。例えば、目的の方向に構造遺伝子が導入されたプラスミドと、逆方向に構造遺伝子が導入されたプラスミドとの割合は、1:1程度である。
【0029】前記構造遺伝子がクローニングされたプラスミドのうち、pUB110に由来する領域を、例えば、制限酵素SphIとEcoRIなどのように目的遺伝子を導入可能な制限酵素で切出す。一方、上記と同じ制限酵素、例えば、SphIとEcoRIとでpUB110を切断し、前記切出したDNA断片と連結し、宿主であるバチルス属細菌を形質転換する。得られた形質転換株の殆どは、目的とするペプチド又は蛋白質を多量に生産できる方向に連結されたプラスミドを保有する。
【0030】前記のようにして得られた高発現ベクターにより、宿主バチルス属細菌を形質転換することにより、ペプチド又は蛋白質を多量に生産する微生物が得られる。前記ベクターによるバチルス属細菌の形質転換は、慣用の方法、例えば、チャン(Chang) らのプロトプラスト化法[Chang, S., et al., Mol. Gen. Genet., 168, 111-115 (1979)]などを利用して行なうことができる。
【0031】前記宿主微生物は、バチルス(Bacillus)属の微生物であり、その菌株は、遺伝子産物が安定である限り特に制限されない。好ましい宿主は、安全性が高く、菌体外にペプチド又は蛋白質を多量に分泌するバチルス・ズブチリス(Bacillussubtilis)である。さらに、バチルス・ズブチリスの中で、突然変異などの手法により菌体外へのプロテアーゼ活性を低下させた菌株が好ましい。このような菌株をプラスミドにより形質転換する場合には、分子量の小さなペプチドであっても、宿主由来のプロテアーゼによる分解を抑制できる。
【0032】プロテアーゼ活性の低い枯草菌としては、例えば、前記バチルス・サブチリス104HL株;バチルス・サブチリスDY−16株(微工研菌寄第9488号);アルカリプロテアーゼ及び中性プロテアーゼの生産能を欠き、かつプロテアーゼ活性が野生株の3%以下である枯草菌に、spo OA△677 変異遺伝子を導入した菌株などが挙げられる。特に好ましい菌株には、例えば、特願平1−281440号において、本出願人が提案したように、バチルス・サブチリス104HL株にspo OA△677 変異遺伝子を導入したバチルス・サブチリスSP011株(微工研菌寄第10987号)、バチルス・サブチリスDY−16株にspo OA△677 変異遺伝子を導入したバチルス・サブチリスSPL14株(微工研菌寄第10988号)などが含まれる。
【0033】特にバチルス・サブチリスSPL14株は、野生株に比べてプロテアーゼ活性が1/5000以下であり、生産物の安定性が極めて良好であること、アミノ酸要求性を2種類有し、胞子を形成しないため、自然界における生育が不可能と考えられることから、実験室における遺伝子組換えだけでなく、有用生産物の工業生産のための宿主として極めて有用な菌である。
【0034】なお、spo OA変異は、胞子形成の最も初期の段階において胞子形成を遮断する突然変異である。この変異遺伝子は、枯草菌が熱、紫外線、化学薬品及び乾燥などに晒された場合、通常自然に生存する形態である胞子を生成する能力を破壊する。この突然変異を起す具体的な方法として、KNO3 をN源とする培地で、対数増殖期末期に達した枯草菌の細胞を繰り返し継代培養する方法が知られている(別冊蛋白質核酸酵素、細菌ファージ遺伝実験法、第213 頁 (1972) )。
【0035】前記ペプチド又は蛋白質は、前記形質転換されたバチルス属細菌を培養することにより得られる。形質転換されたバチルス属細菌の培養は、慣用の液体培養に準じて行なうことができる。すなわち、形質転換株の培養は、慣用の成分、例えば、無機塩、炭素源、窒素源、増殖因子成分などを含む液体培地で、振盪培養又は通気撹拌培養法により行なうことができる。培地のpHは、例えば、7〜8程度である。培養は、微生物の培養に採用される通常の条件、例えば、温度15〜45℃、好ましくは25〜40℃、培養時間6〜60時間程度の条件で行なうことができる。
【0036】なお、宿主微生物が生産したペプチド又は蛋白質の分離精製は、慣用の方法、例えば、塩析、溶媒沈澱法、透析法、限外濾過法、ゲル濾過法、SDS−アガロースゲル電気泳動法、SDS−ポリアクリルアミドゲル電気泳動法、電気溶出(electro elution) 法、イオン交換クロマトグラフィー法、アフィニティークロマトグラフィー法、逆相高速液体クロマトグラフィー法や、これらを組合せた方法で行なうことができる。ペプチドなどが菌体内やペリプラズムに蓄積される場合には、慣用の方法、例えば、菌体を破砕し、前記分離精製に供することにより、ペプチド又は蛋白質を得ることができる。
【0037】
【実施例】以下に、実施例に基づいて本発明をより詳細に説明するが、本発明はその要旨を越えない限り、以下の実施例により限定されるものではない。
【0038】実施例1アッセイ系としてα−アミラーゼ遺伝子を選び、分泌生産されるα−アミラーゼの量を指標として、高発現の分泌ベクターを構築した。
(1)バチルス・サブチリスSPL14株の作製spo OA遺伝子及びリンコマイシン耐性遺伝子(以下、Linr と称する)を有する枯草菌として知られているバチルス・サブチリスATCC39096株を、表1に示すLB培地を用いて37℃で対数増殖期まで振盪培養した。
【0039】
【表1】

培養液50ml中の菌体を集め、斉藤・三浦の方法[H. Saito, K. Miura, Biochem. Biophis. Acta, 72, 619 (1963)]により染色体DNAを抽出精製した。
【0040】得られた染色体DNAを用いてコンピテントセル形質転換法によりバチルス・サブチリスDY−16株を形質転換した。形質転換した枯草菌を、ヒスチジンを50mg/l含む最小寒天培地にまき、37℃で48時間培養した。胞子形成能欠損株を、以下の指標に従って選択した。
【0041】1.胞子形成培地に植菌し、そのコロニーがメラニン色素生産能を失ったこと2.80℃、10分の熱処理に耐性を示すこと3.顕微鏡検査により胞子形成を認めないこと以下の方法でプロテアーゼ活性を測定し、得られた胞子欠損株173株のうち最もプロテアーゼ活性の低下した株を選出した。
【0042】先ず、1%カゼインを含むLB寒天平板培地に、それぞれの胞子欠損形質転換株と、バチルス・サブチリスDY−16株を別々に植菌し、37℃で24時間培養した。菌体が分泌するプロテアーゼによりカゼインが分解されるので、カゼインの分解に伴なって形成される菌体の周りのハローの大きさを指標として、親株であるバチルス・サブチリスDY−16株よりもプロテアーゼ生産能が低下した40株を分離した。
【0043】さらに、これらの株をLB培地で液体培養し、その培養上清に存在するプロテアーゼ活性を、FITC−カゼイン(ジクマ社)を基質として測定した。すなわち、バチルス・ズブチリスDY−16及びプロテアーゼ活性が低下した形質転換株40株をそれぞれ、LB培地10mlを用いて一晩培養し、その培養液1mlを遠心分離し、その上清をプロテアーゼ酵素液とした。
【0044】酵素液50μlと、0.2μgのFITC−カゼインとを、10mMのトリス−塩酸(pH7.5)および2mMのCaCl2 の緩衝液中(200μl)で37℃で2時間反応した。反応液に200μlの7.5%トリクロロ酢酸を添加し、遠心分離した後、その上清100μlに、500mMトリス−塩酸(PH8.5)900μlを添加し、プロテアーゼ活性を、螢光光度計を用い、波長600nmでの光学濃度(OD600 )により測定した。
【0045】なお、プロテアーゼ活性(PUD)は、37℃、pH7.5において、カゼインから1分間に1μモルのチロシンTyrに相当するトリクロロ酢酸可溶性ペプチドを遊離する酵素量を1PUDとして、算出した。その結果、プロテアーゼ活性が親株バチルス・サブチリスDY−16株に比べ極端に低下した4株(バチルス・サブチリスSPL9,11,14,39)を得た。
【0046】これら4株のプロテアーゼ活性をさらに詳しく測定した。バチルス・サブチリスDY−16株及びプロテアーゼ活性が低下した形質転換株4株を10mlのLB培地で18時間培養した後、50mlのMedium A培地に菌体を移し、培養24時間及び48時間後の培養上清中のプロテアーゼ活性を測定した。その結果を表2に示す。
【0047】
【表2】
得られたこれらの株は、親株に比べプロテアーゼ活性が1/20以下と低いことが確認された。特にバチルス・サブチリスSPL14株は、プロテアーゼ活性が低い。また、いずれの株においてもヒスチジンとロイシンに対する要求性を有しており、遺伝子組換え実験における宿主として要求される複数の栄養要求性を確認した。これらの菌株は、ペプチドを効率よく生産できる宿主として、安全かつ有効に用いることができる。
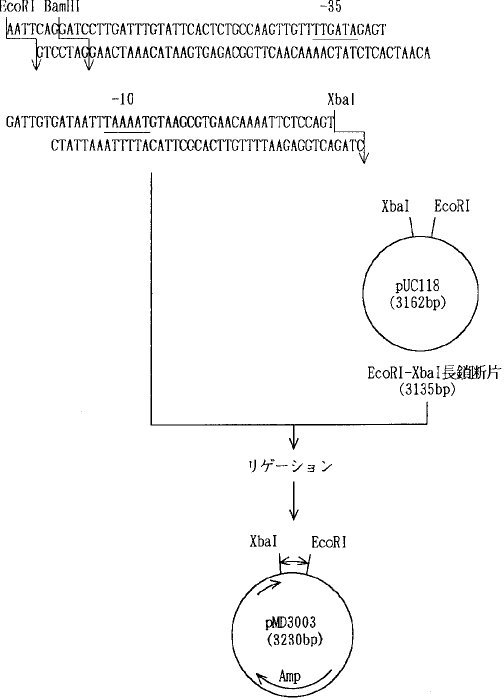
(2)α−アミラーゼ遺伝子のプローモーターの合成以下のような枯草菌α−アミラーゼ遺伝子のプローモーター配列をそれぞれ化学合成し、両遺伝子をアニーリングした後、pUC118のEcoRI−XbaIの間に挿入し、図2に示すようにプラスミドpMD3003を構築した。
【0048】
5'- AATTCAGGATCCTTGATTTGTATTCACTCTGCCAAGTTGTTTTGATAGAGTGATTGTGATAATTTAAAATGTAAGCGTGAACAAAATTCTCCAGT-3'5'-ACTGGAGAATTTTGTTCACGCTTACATTTTAAATTATCACAATCACTCTATCAAAACAACTTGGCAGAGTGAATACAAATCAAGATCCTG-3'(3)SD配列の合成枯草菌のα−アミラーゼ遺伝子及び黄色ブドウ球菌由来のプロテインA遺伝子のSD配列、及び枯草菌16SリボソームRNA(rRNA)の配列を参考として、以下の塩基配列で表されるSD配列を、ABI社DNA合成装置を用いて合成した。なお、遺伝子の5′側にはプローモーターとの連結部位であるXbaI部位を、3′領域にはα−アミラーゼ遺伝子のシグナル配列との連結部位であるNruI部位を導入した。
【0049】CTAGAAAGGAGGTGATCTAGAATTCATGTTCGTTTCCTCCACTACATCTTAAGTACAAGC(4)α−アミラーゼ遺伝子バチルス・サブチリス1A412株[オハイオ州立大学のバチルス・ジェネティク・ストック・センター(Bacillus Genetic Stock Center)(BGSC)より入手した]から前記斉藤・三浦の方法により調製した染色体DNA10μgを、100μlのSau3AI溶液[10mMのトリス−塩酸(pH7.5)、7mMのMgCl2 、100mMのNaCl)に溶解し、制限酵素Sau3AIを1単位加え、37℃で10分間反応させ、フェノール溶液[フェノールをTE緩衝液(10mMのトリス−塩酸(pH8.0)、1mMのEDTA)で飽和したもの]を添加し、反応を停止した後、エタノール沈澱物として、枯草菌染色体DNAのSau3AIの部分分解物を得た。
【0050】一方、1μgの枯草菌ベクターpUB110を、BamHI溶液[10mMのトリス−塩酸(pH8.0)、7mMのMgCl2 、100mMのNaCl、2mMの2−メルカプトエタノール、0.01%の牛血清アルブミン(BSA)]中で5単位の制限酵素BamHIと37℃で1時間反応させ、フェノール溶液で処理した後、エタノール沈澱物をpUB110のBamHI分解物として得た。
【0051】次いで、得られた上記二つのDNA断片のリゲーションを行った。すなわち、上記のようにして調製したそれぞれのDNA断片を、10μlのリゲーション溶液[66mMのトリス−塩酸(pH7.6)、6.6mMのMgCl2 、10mMのジチオスレイトール(DTT)、および0.1mMのアデノシン三リン酸(ATP)]に溶解した後、混合し、T4 DNAリガーゼ10単位を添加し、20℃で1時間反応を行った。
【0052】このDNA溶液を用いて、バチルス・サブチリス1A289株(BGSCより入手)をプロトプラスト化法(Chan, S., Cohen, N., Molec, gen. Genet., 168, 111-115 (1979) )により形質転換した。
【0053】形質転換株を寒天培地で培養し、カナマイシン耐性を示す約2万個のコロニーが得られ、寒天平板培地上でのヨード・デンプン反応により、このうち1株が菌体外にα−アミラーゼを分泌生産していることが確認された。
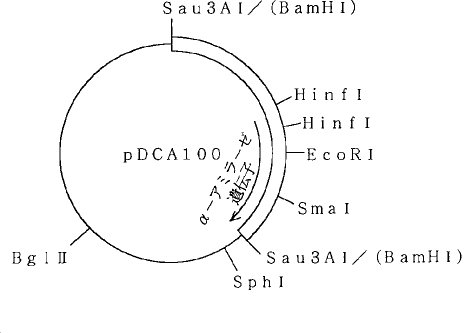
【0054】この株からプラスミドDNAを分離し、制限酵素による解析を行なったところ、図3に示すような制限酵素地図が得られ、前記プラスミドをpDCA100と命名した。pDCA100は、pUB110のBamHI部位に3.1KbのSau3AI断片が挿入された7.6Kbのプラスミドである。前記3.1KbのSau3AI断片の中には、枯草菌由来のα−アミラーゼ遺伝子[Yamazaki, H.et al., J. Bacteriol., 156 , 327 (1984)]と全く同じ制限酵素パターンが認められたことから、α−アミラーゼ遺伝子がクローニングされていることが確認された。
【0055】pDCA100のα−アミラーゼ遺伝子と同一と考えられるα−アミラーゼ遺伝子が導入された菌株は、アメリカ合衆国オハイオ大学バチルス・ジェネテック・ストック・センター(BGSC)にECE35株として登録されている。
(5)α−アミラーゼ遺伝子とSD配列の連結クローニングされたα−アミラーゼ遺伝子と合成SD配列とを、以下の塩基配列で表される合成遺伝子を介して連結した。
【0056】
5'-CGAAACGATTCAAAACCTCTTTACTGCCGTTATTTGCTGGATTTTTATTGCTGTTTTATTTGGTTCTGGCAGGAC-3'5'-CGGTCCTGCCAGAACCAAATAAAAACAGCAATAAAAATCCAGCGAATAACGGCAGTAAAGAGGTTTTGAATCGTTTTCG-3'この合成遺伝子は、α−アミラーゼ遺伝子のシグナル配列部分のアミノ酸配列(Yamazaki, H. et al., J. Bacteriol., 156 , 327 (1984))に従って塩基配列を決定し、化学合成した。常法に従って精製した後、アニーリングし、合成DNAリンカーとした。
(6)ターミネーターの作製枯草菌のアルカリプロテアーゼ遺伝子のターミネーターを参考として、以下のターミネーターをそれぞれ合成した。なお、5′末端側には、BglII部位を、3′末端側にはSphI部位を設けた。また、3′末端のすぐ近くにはBamHI部位を設けた。合成した後、両合成遺伝子をアニーリングし、ターミネーター断片とした。
【0057】
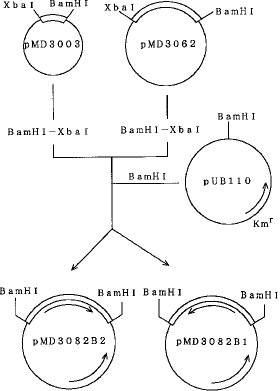
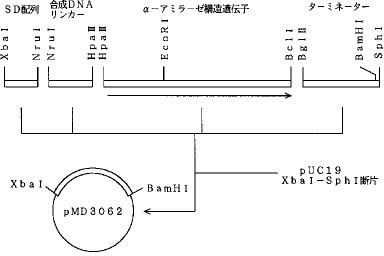
5'-GATCTATAAAAAGAAGCAGGTTCCTCCATACCTGCTTCTTTTTATTTGTCAGCATCCTGATGTTGGATCCGCATG-3'5'-CGGATCCAACATCAGGATGCTGACAAATAAAAAGAAGCAGGTATGGTGGAACCTGCTTCTTTTTATA-3'(7)SD配列、α−アミラーゼ遺伝子、ターミネーターの連結SD配列、α−アミラーゼ遺伝子、ターミネーターを、図4に示すように連結し、プラスミドpMD3062を得た。すなわち、DNA合成したSD配列、リンカーDNA、ターミネーター断片をDNAキナーゼを用いてカイネーションした。α−アミラーゼ遺伝子は、pDCA100から、HpaII−EcoRI断片、EcoRI−BclI断片として回収した。これらの遺伝子と、アルカリホスファターゼ処理したpUC19のXbaI−SphI切断断片と、α−アミラーゼ遺伝子部分を混合し、DNAリガーゼを用いて連結し、大腸菌HB101株を形質転換した。得られたアンピシリン耐性形質転換株の中から、目的とするプラスミドpMD3062を得た。
(8)pMD3082B1の構築とα−アミラーゼの生産プラスミドpMD3082B1の構築図を図5に示す。
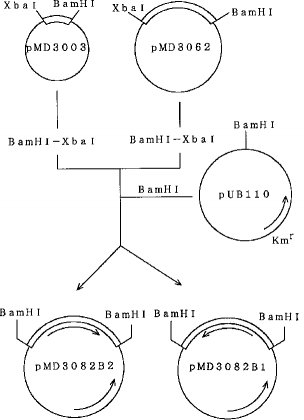
【0058】先ず、pMD3003を制限酵素BamHIとXbaIとで切断し、プローモーターを切り出した。また、pMD3062をXbaIとBamHIとを用いて切断し、SD配列からターミネーターまでの領域を含むα−アミラーゼ遺伝子部分を得た。そして、これらの遺伝子を、アルカリホスファターゼ処理したpUB110のBamHI消化物と混合し、DNAリガーゼを用いて連結し、バチルス・サブチリスSPL14株をプロトプラスト化法により形質転換した。
【0059】得られたカナマイシン耐性形質転換株の中から、α−アミラーゼ生産能を示す200株の形質転換株を分離した。これらの形質転換株の中から、α−アミラーゼ遺伝子の方向性の異なる2種類のプラスミドが得られた。pUB110中のカナマイシン耐性遺伝子と同じ方向で遺伝子が導入されたプラスミドをpMD3082B1、その逆方向に導入されたプラスミドをpMD3082B2とした。それらの構成比は前者:後者=1:40であった。
【0060】次いで、これらのプラスミドを有するバチルス・サブチリスSPL14株を12時間浸透培養し、得られたα−アミラーゼ活性を比較検討したところ、pMD3082B1の方がpMD3082B2よりも約3倍のα−アミラーゼを生産し、培養12時間後には2000ユニット/mlのα−アミラーゼを生産した(表3参照)。なお、α−アミラーゼ活性は、不破らの方法(Fuwa, H., J. Biochem, 41, 583-603 (1954))に従って測定した。
【0061】
【表3】
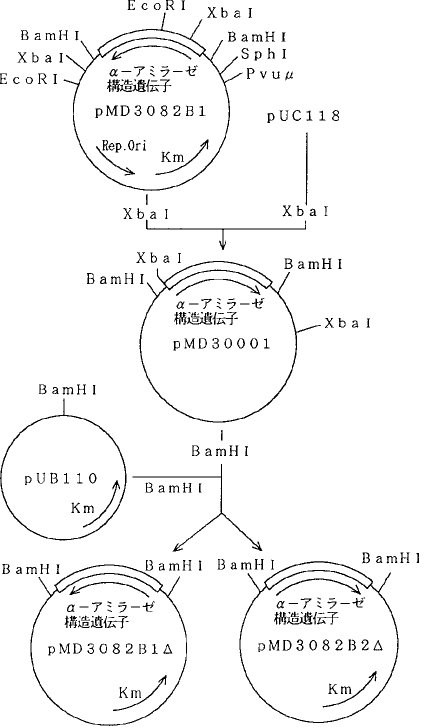
(9)プローモーターを除去したときのα−アミラーゼ活性pMD3062をXbaIで切断し、SD配列、α−アミラーゼ遺伝子、ターミネーター遺伝子部分を回収し、制限酵素XbaIで切断したpUC118のXbaI切断部位に連結した。α−アミラーゼ遺伝子の5′側に、pUC118に存在するポリリンカー部位であるBamHI部位を位置させて連結したプラスミドをpMD30001とした。
【0062】このpMD30001を、制限酵素BamHIにより切断することにより、プローモーターが存在しないα−アミラーゼ遺伝子を回収できる。そこで、pMD3001をBamHIで切断した後、アルカリホスファターゼで処理したpUB110のBamHI切断断片と混合し、DNAリガーゼを用いて連結した後、バチルス・サブチリスSPL14株を形質転換した。得られたカナマイシン耐性形質転換株を培養し、それぞれプラスミドDNAを調製し、α−アミラーゼ遺伝子の方向性を調べたところ、pUB110にα−アミラーゼ遺伝子がカナマイシン耐性遺伝子と同じ方向に導入されたプラスミドpMD3082B1△と、逆方向に導入されたpMD3082B2△を得た。プラスミドpMD3082B1△およびpMD3082B2△の構築図を図6に示す。
【0063】それぞれのプラスミドを保有するバチルス・サブチリスSPL14株を培養し、得られた培養濾液中のα−アミラーゼ活性を調べたところ、表4に示す結果を得た。すなわち、pMD3082B2△を保有するバチルス・サブチリスSPL14株は、培養12時間後にα−アミラーゼを10ユニットしか生産せず、プラスミドを保有していないSPL14株の生産するα−アミラーゼ活性と変りがなかった。一方、pMD3082B1△を有するバチルス・サブチリスSPL14株は、培養12時間後にα−アミラーゼを800ユニット/ml生産した。
【0064】
【表4】
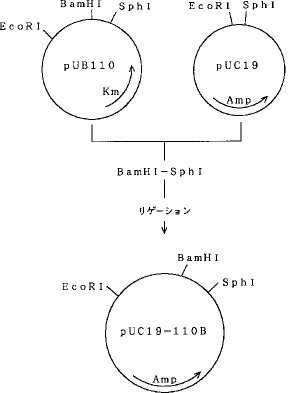
実施例2プラスミドpUB110を制限酵素SphIおよびEcoRIで切断し、SphI部位からEcoRI部位までの小さな約1Kbpの断片を得た。プラスミドpUC19を制限酵素SphIおよびEcoRIで切断し、pUC19のポリリンカーサイトに、DNAリガーゼを用いて前記約1Kbpの断片を導入し、プラスミドpUC19−110Bを構築した。プラスミドpUC19−110Bの構築図を図7に示す。
【0065】このプラスミドベクター上のpUB110部分に存在するBamHI部位を酵素BamHIにより解裂し、アルカリホスファターゼ処理した。また、前記pMD3082B1からα−アミラーゼ遺伝子部分を制限酵素BamHIで切断し回収した。両遺伝子断片を混合し、リガーゼにより連結し、大腸菌HB101株を形質転換した。
【0066】得られたアンピシリン耐性を有する形質転換株から、プラスミドを調製し、遺伝子の導入方向を調べたところ、供試48株のうち23株が、目的の方向に遺伝子が導入されたプラスミドを有していた。
【0067】得られたプラスミドを、制限酵素SphIとEcoRIとで切断し、DNA断片を調製した。また、pUB110を制限酵素SphIとEcoRIとで切断し、DNAリガーゼを用いて、前記DNA断片と連結することにより、遺伝子を、目的のカナマイシン耐性遺伝子と同じ方向に100%導入できた。
【図面の簡単な説明】
【図1】ベクタープラスミドpUB110の概略制限酵素地図である。
【図2】プラスミドpMD3003の構築図である。
【図3】プラスミドpDCA100の制限酵素地図である。
【図4】プラスミドpMD3062の構築図である。
【図5】プラスミドpMD3082B1の構築図である。
【図6】プラスミドpMD3082B1△及びpMD3082B2△の構築図である。
【図7】プラスミドpUC19−110Bの構築図である。
【0001】
【産業上の利用分野】本発明は、枯草菌を代表とするバチルス属細菌で利用可能な新規な高発現ベクター、このベクターを保有するバチルス属細菌、およびこのバチルス属細菌によるペプチド又は蛋白質の製造方法に関する。
【0002】
【従来の技術と発明が解決しようとする課題】枯草菌(Bacillus subtilis)を代表とするバチルス(Bacillus)属細菌は、古くから種々の菌体外酵素の生産菌として、工業的に利用されている。例えば、枯草菌の仲間は、日本において古くから納豆の製造に使用されており、大腸菌などで問題となるエンドトキシンを生産しないこと、人体に寄生しないことなどの理由から、極めて安全性の高い微生物である。このように、枯草菌は、ペプチド又は蛋白質の工業的な生産菌として、優れた性質を有し、その有用性が注目されている。例えば、特開昭56−137986号公報には、前記菌体外酵素の遺伝子のプロモーター、SD配列(シャイン−ダルガーノ(Shine-Dalgarno)配列)、シグナル遺伝子をコードするDNAをクローニングし、その下流に目的とするペプチド又は蛋白質をコードする遺伝子を連結し、これを枯草菌に導入し、目的とするペプチド又は蛋白質を菌体外に分泌生産させている。
【0003】しかしながら、枯草菌におけるペプチド又は蛋白質の生産は、枯草菌自体が産生するα−アミラーゼやプロテアーゼなどが主である。すなわち、枯草菌が高い分泌生産能を有するにも拘らず、他の微生物由来の酵素及び比較的分子量の小さな哺乳類の生理活性ペプチドなどを生産させると、菌体自体が菌体外に多量に分泌する各種のプロテアーゼにより生産物が分解され、高い収率で有用生産物を回収できない。
【0004】そこで、宿主枯草菌のプロテアーゼ活性を低下させるための研究がなされている。例えば、プロテアーゼ活性の低い枯草菌として、枯草菌の主たる菌体外プロテアーゼである中性プロテアーゼ及びアルカリプロテアーゼの両者を欠失したバチルス・サブチリス(Bacillus subtilis)104HL株[バイオケミカル・アンド・バイオフィジカル・リサーチ・コミュニケーションズ(Biochem. Biophys.Res. Commun.), 128; 601-606, 1985]が知られている。また、本出願人は、特開昭64−37284号公報において、バチルス・サブチリス104HL株に、pap 遺伝子を導入したバチルス・サブチリスDY−16株(微工研菌寄第9488号)を提案した。
【0005】一方、枯草菌ベクターの開発も、宿主の開発と同様に古くから行なわれている。例えば、枯草菌自体や、バチルス・アミロリクエファシエンス、バチルス・リケニホルミスなどの類縁菌のα−アミラーゼ、または中性及びアルカリプロテアーゼの遺伝子のプロモーター、リボソーム結合領域(SD配列)、シグナル配列を利用した分泌ベクターが発表されている。これらの分泌ベクターにより、それぞれ本来のα−アミラーゼやプロテアーゼの分泌だけでなく、例えばプロテインAなどの他の有用酵素、およびインターフェロン、ヒト成長ホルモンなどの生理活性ペプチドを生産できる。
【0006】しかしながら、枯草菌が分泌生産という有用性を有するにも拘らず、大腸菌に比べて、遺伝子組換えの宿主としての利用が遅れている。このことは、遺伝子産物の生産量が少ないことに起因する。すなわち、大腸菌系では、多くの強いプロモーター系と、極めて正確な制御系とが確立され、これらが多コピープラスミド上で安定に機能している。これに対して、枯草菌の系においては、多くの試みにも拘らず、大腸菌のように確立された系は、存在しない。
【0007】
【発明が解決しようとする課題】従って、本発明の目的は、バチルス属細菌内に安定に保持され、かつペプチド又は蛋白質を菌体外に多量に分泌生産させることができる高発現ベクターを提供することにある。
【0008】本発明の他の目的は、前記高発現ベクターを保有し、ペプチド又は蛋白質を菌体外に多量に分泌生産するバチルス属細菌を提供することにある。
【0009】本発明のさらに他の目的は、ペプチド又は蛋白質を効率よく多量に得ることができるペプチド又は蛋白質の製造方法を提供することにある。
【0010】
【発明の構成】本発明者らは、高い分泌能を有するバチルス属細菌に着目し、その分泌ベクターの開発を鋭意研究した結果、枯草菌において一般に使用されているベクタープラスミドpUB110のクローニングサイトBamHI切断部位またはその近傍に、高い翻訳能力を有する遺伝子を特定の方向に導入すると、分泌ベクターが少なくともバチルス属細菌内で安定に保持され、かつ分泌ベクターにより所望の遺伝子産物が多量に分泌発現することを見いだし、本発明を完成した。
【0011】すなわち、本発明は、少なくともpUB110のSphI切断部位からBamHI切断部位までの塩基配列を含むpUB110由来のベクターであって、BamHI切断部位またはその近傍に、SphI切断部位からBamHI切断部位への方向に、SD配列を有するペプチド又は蛋白質をコードする遺伝子が連結されている高発現ベクターを提供する。
【0012】本発明は、前記高発現ベクターを保有するバチルス属細菌を提供する。
【0013】さらに本発明は、前記バチルス属細菌を培養し、産生したペプチド又は蛋白質を回収するペプチド又は蛋白質の製造方法を提供する。
【0014】本明細書において、塩基配列における核酸の表示はIUPACにより採択されている表示法に従うものとする。
【0015】本発明の高発現ベクターは、ベクタープラスミドpUB110に由来し、pUB110のSphI切断部位からBamHI切断部位までの塩基配列を含んでいる。pUB110はその全塩基配列を有している必要はなく、少なくともSphIおよびBamHI切断部位を有していればよく、SphI、BamHIおよびEcoRI切断部位を有するpUB110由来のベクターであってもよい。なお、pUB110は黄色ブドウ状球菌[スタフィロコッカス(Staphylococcus ) ]属に由来する多コピープラスミドであり、カナマイシン耐性遺伝子を有し、その制限地図は確立されている。図1にベクタープラスミドpUB110の制限酵素地図の概要を示す。
【0016】pUB110とペプチド又は蛋白質をコードする遺伝子との連結部位は、制限酵素BamHI部位に限らず、その近傍、例えば、BamHI部位からEcoRI部位までの間などであってもよい。すなわち、EcoRI部位や、BamHI部位からEcoRI部位までの領域に存在するAvaI部位、XbaI部位に、ペプチド又は蛋白質をコードする遺伝子を連結してもよい。
【0017】クローニングサイトBamHI切断部位またはその近傍には、高い翻訳能力を有する遺伝子、すなわちSD配列を有するペプチドまたは蛋白質をコードする遺伝子が連結されている。
【0018】本発明の主たる特徴は、前記ペプチド又は蛋白質をコードする遺伝子が、従来とは逆の方向であるSphI切断部位からBamHI切断部位への方向、すなわちカナマイシン耐性遺伝子と同じ方向に導入されている点にある。このような特定の方向にペプチド又は蛋白質をコードする構造遺伝子を導入すると、宿主微生物により、ペプチド又は蛋白質を多量に分泌生産させることができる。
【0019】前記ペプチド又は蛋白質は、特に限定されず、酵素、原核細胞および真核細胞を問わず、動物性細胞由来のペプチド又は蛋白質であってもよい。ペプチド及び蛋白質には、例えば、インスリン、ガストリン、各種オピオノイドペプチド、上皮細胞成長因子、エンドセリン、バソアクティブ・インテスティナル・ポリペプチド[Vasoactive Intestinal Polypeptide (VIP)]、心房性利尿ホルモン(ANP)、サブスタンスP、カルシトニン、インシュリン様成長因子I,II、ガラニン、モチリン、バソプレシン、インターフェロン、IL−2などの各種の生理活性ペプチドまたはこれらの前駆体;α−アミラーゼ、プロテインA、ヒルジン、エグリンC、分泌性白血球由来プロテアーゼインヒビターなどの阻害剤、ヒトアルブミン、血液凝固因子、リンフォカイン、神経成長因子、肝細胞再生因子などの各種の分化誘導因子、成長因子などの蛋白質またはこれらの前駆体が含まれる。
【0020】前記SD配列は、特に制限されず、例えば、枯草菌の16S−rRNAの配列に対応して合成した下記のSD配列であってもよく、5′−CTAGAAAGGAGGTGATCTAGAATTC下記のSD配列などであってもよい。
【0021】5′−CTAGAAAGGAGGTGATAGAATTC5′−CTAGAAAGGAGGTGAGAATTC5′−CTAGAAAGGAGGTGAGAATAT5′−CTAGAAAGGAGGTGAGAATこれらのSD配列を導入すると、僅の発現の差異が認められるものの、ベクタープラスミドによる効果が本質的に損われるものではない。SD配列は、リボソーム結合部位として機能し、その下流には、開始コドンが存在する。
【0022】なお、従来、pUB110のBamHI切断部位が関与して、何かしらの転写が行なわれていることが知られていた[Bruchner, R.,et al., Gene 32, 151-160 (1984)およびCurran, J.F., and Stewart, C.J., Virology, 142, 98-111 (1985) ]。しかしながら、この知見を積極的に利用した例はない。このことは、各種のファクターが転写の強さについて及ぼす影響を正確に定量化できていなかったことに他ならない。
【0023】前記ペプチド又は蛋白質をコードする遺伝子を、SD配列と共に、ベクターDNAに前記特定の方向に連結することにより、プロモーターを導入しなくても、SD配列の強さに応じてペプチド又は蛋白質を生産できる。従って、強いSD配列であれば、プロモーターは必ずしも必要ではないが、SD配列の5′側にプロモーターを連結するのが好ましい。プロモーターとしては、例えば、枯草菌のα−アミラーゼ遺伝子のプロモーターなどが挙げられる。
【0024】構造遺伝子の下流、すなわちペプチド又は蛋白質をコードする遺伝子の3′側に、ターミネーターが連結されていなくても、ベクターによる効果が特に損われることはないが、構造遺伝子の下流には、ターミネーターが連結されているのが好ましい。ターミネーターは、特に限定されず、例えば、枯草菌のアルカリプロテアーゼ遺伝子のターミネーターや、これに対応して化学合成したターミネーターなどであってもよい。
【0025】本発明の高発現ベクターおよびこのベクターを保有するバチルス属細菌は、種々の方法で構築できる。例えば、pUB110を制限酵素BamHIで解裂し、アルカリホスファターゼで処理する。また、所望のペプチド又は蛋白質をコードする遺伝子の5′側にプロモーターおよびSD配列を、3′側にターミネーターを連結し、DNA断片を調製する。このDNA断片をpUB110のBamHI部位に導入する。なお、前記DNA断片とBamHI部位とのサイトが合わない場合には、両断片を平滑末端としたり、適当な制限酵素リンカーを用いればよい。pUB110にペプチド又は蛋白質をコードする遺伝子が導入されたキメラプラスミドにより、宿主微生物であるバチルス属細菌を形質転換する。得られた形質転換株から、カナマイシン耐性遺伝子と同じ方向に前記遺伝子が導入されたプラスミドを選択することにより、前記高発現ベクターを保有するバチルス属細菌が得られる。
【0026】しかし、この方法では、前記制限酵素による切断部位が、目的とするペプチド又は蛋白質ペプチドの遺伝子に存在しない場合には、pUB110のBamHI部位に構造遺伝子を両方向に導入し、得られたキメラプラスミドの中から、目的とするベクターを捜し出す必要がある。この方法では、目的蛋白質が枯草菌由来のα−アミラーゼであっても、カナマイシン耐性遺伝子と同じ方向に遺伝子が導入されたプラスミドを得る確率は、通常、比較的小さい。すなわち、枯草菌由来の構造遺伝子をカナマイシン耐性遺伝子と同じ方向に導入されたプラスミドは、逆方向に導入されたプラスミドに比べて、例えば、1/40程度しか得られない。さらに、枯草菌以外のペプチド又は蛋白質をコードする遺伝子を導入すると、枯草菌の生育阻害が顕著となり、目的とするプラスミドが得られにくくなる。このことを解決するためには、例えば、次の2つの方法が採用できる。
【0027】第1の方法では、pUB110をBamHIおよびEcoRIで切断し、アルカリホスファターゼで処理する。一方、所望のペプチド又は蛋白質をコードする遺伝子の5′側をBamHI部位、3′側をEcoRI部位となるように切断するか、又は合成DNAリンカーを用いてDNA断片を調製する。そして、pUB110のBamHIとEcoRI部位の間に、前記DNA断片を方向性をもって導入する。なお、この方法において、pUB110を制限酵素BamHIとXbaI、制限酵素XbaIとEcoRIなどで切断し、上記と同様にして、目的の遺伝子を方向性を考慮して導入してもよい。
【0028】第2の方法では、先ずpUB110を制限酵素SphIとEcoRIとで切断し、得られた約1キロベースペア(Kbp)の遺伝子断片を、大腸菌のベクター、例えば、プラスミドpUC19などのポリリンカーサイトEcoRI−SphI間に導入する。得られたプラスミドのpUB110に由来する遺伝子のうち、例えば、BamHI部位に、前記ペプチド又は蛋白質をコードする遺伝子を導入し、宿主大腸菌、例えばHB101などを形質転換する。得られた形質転換株からプラスミドを調製し、目的の方向に遺伝子が導入されたプラスミドを選択する。この際、pUB110上に、カナマイシン耐性遺伝子と同じ方向に構造遺伝子が導入されたプラスミドが多量に得られる。例えば、目的の方向に構造遺伝子が導入されたプラスミドと、逆方向に構造遺伝子が導入されたプラスミドとの割合は、1:1程度である。
【0029】前記構造遺伝子がクローニングされたプラスミドのうち、pUB110に由来する領域を、例えば、制限酵素SphIとEcoRIなどのように目的遺伝子を導入可能な制限酵素で切出す。一方、上記と同じ制限酵素、例えば、SphIとEcoRIとでpUB110を切断し、前記切出したDNA断片と連結し、宿主であるバチルス属細菌を形質転換する。得られた形質転換株の殆どは、目的とするペプチド又は蛋白質を多量に生産できる方向に連結されたプラスミドを保有する。
【0030】前記のようにして得られた高発現ベクターにより、宿主バチルス属細菌を形質転換することにより、ペプチド又は蛋白質を多量に生産する微生物が得られる。前記ベクターによるバチルス属細菌の形質転換は、慣用の方法、例えば、チャン(Chang) らのプロトプラスト化法[Chang, S., et al., Mol. Gen. Genet., 168, 111-115 (1979)]などを利用して行なうことができる。
【0031】前記宿主微生物は、バチルス(Bacillus)属の微生物であり、その菌株は、遺伝子産物が安定である限り特に制限されない。好ましい宿主は、安全性が高く、菌体外にペプチド又は蛋白質を多量に分泌するバチルス・ズブチリス(Bacillussubtilis)である。さらに、バチルス・ズブチリスの中で、突然変異などの手法により菌体外へのプロテアーゼ活性を低下させた菌株が好ましい。このような菌株をプラスミドにより形質転換する場合には、分子量の小さなペプチドであっても、宿主由来のプロテアーゼによる分解を抑制できる。
【0032】プロテアーゼ活性の低い枯草菌としては、例えば、前記バチルス・サブチリス104HL株;バチルス・サブチリスDY−16株(微工研菌寄第9488号);アルカリプロテアーゼ及び中性プロテアーゼの生産能を欠き、かつプロテアーゼ活性が野生株の3%以下である枯草菌に、spo OA△677 変異遺伝子を導入した菌株などが挙げられる。特に好ましい菌株には、例えば、特願平1−281440号において、本出願人が提案したように、バチルス・サブチリス104HL株にspo OA△677 変異遺伝子を導入したバチルス・サブチリスSP011株(微工研菌寄第10987号)、バチルス・サブチリスDY−16株にspo OA△677 変異遺伝子を導入したバチルス・サブチリスSPL14株(微工研菌寄第10988号)などが含まれる。
【0033】特にバチルス・サブチリスSPL14株は、野生株に比べてプロテアーゼ活性が1/5000以下であり、生産物の安定性が極めて良好であること、アミノ酸要求性を2種類有し、胞子を形成しないため、自然界における生育が不可能と考えられることから、実験室における遺伝子組換えだけでなく、有用生産物の工業生産のための宿主として極めて有用な菌である。
【0034】なお、spo OA変異は、胞子形成の最も初期の段階において胞子形成を遮断する突然変異である。この変異遺伝子は、枯草菌が熱、紫外線、化学薬品及び乾燥などに晒された場合、通常自然に生存する形態である胞子を生成する能力を破壊する。この突然変異を起す具体的な方法として、KNO3 をN源とする培地で、対数増殖期末期に達した枯草菌の細胞を繰り返し継代培養する方法が知られている(別冊蛋白質核酸酵素、細菌ファージ遺伝実験法、第213 頁 (1972) )。
【0035】前記ペプチド又は蛋白質は、前記形質転換されたバチルス属細菌を培養することにより得られる。形質転換されたバチルス属細菌の培養は、慣用の液体培養に準じて行なうことができる。すなわち、形質転換株の培養は、慣用の成分、例えば、無機塩、炭素源、窒素源、増殖因子成分などを含む液体培地で、振盪培養又は通気撹拌培養法により行なうことができる。培地のpHは、例えば、7〜8程度である。培養は、微生物の培養に採用される通常の条件、例えば、温度15〜45℃、好ましくは25〜40℃、培養時間6〜60時間程度の条件で行なうことができる。
【0036】なお、宿主微生物が生産したペプチド又は蛋白質の分離精製は、慣用の方法、例えば、塩析、溶媒沈澱法、透析法、限外濾過法、ゲル濾過法、SDS−アガロースゲル電気泳動法、SDS−ポリアクリルアミドゲル電気泳動法、電気溶出(electro elution) 法、イオン交換クロマトグラフィー法、アフィニティークロマトグラフィー法、逆相高速液体クロマトグラフィー法や、これらを組合せた方法で行なうことができる。ペプチドなどが菌体内やペリプラズムに蓄積される場合には、慣用の方法、例えば、菌体を破砕し、前記分離精製に供することにより、ペプチド又は蛋白質を得ることができる。
【0037】
【実施例】以下に、実施例に基づいて本発明をより詳細に説明するが、本発明はその要旨を越えない限り、以下の実施例により限定されるものではない。
【0038】実施例1アッセイ系としてα−アミラーゼ遺伝子を選び、分泌生産されるα−アミラーゼの量を指標として、高発現の分泌ベクターを構築した。
(1)バチルス・サブチリスSPL14株の作製spo OA遺伝子及びリンコマイシン耐性遺伝子(以下、Linr と称する)を有する枯草菌として知られているバチルス・サブチリスATCC39096株を、表1に示すLB培地を用いて37℃で対数増殖期まで振盪培養した。
【0039】
【表1】
培養液50ml中の菌体を集め、斉藤・三浦の方法[H. Saito, K. Miura, Biochem. Biophis. Acta, 72, 619 (1963)]により染色体DNAを抽出精製した。
【0040】得られた染色体DNAを用いてコンピテントセル形質転換法によりバチルス・サブチリスDY−16株を形質転換した。形質転換した枯草菌を、ヒスチジンを50mg/l含む最小寒天培地にまき、37℃で48時間培養した。胞子形成能欠損株を、以下の指標に従って選択した。
【0041】1.胞子形成培地に植菌し、そのコロニーがメラニン色素生産能を失ったこと2.80℃、10分の熱処理に耐性を示すこと3.顕微鏡検査により胞子形成を認めないこと以下の方法でプロテアーゼ活性を測定し、得られた胞子欠損株173株のうち最もプロテアーゼ活性の低下した株を選出した。
【0042】先ず、1%カゼインを含むLB寒天平板培地に、それぞれの胞子欠損形質転換株と、バチルス・サブチリスDY−16株を別々に植菌し、37℃で24時間培養した。菌体が分泌するプロテアーゼによりカゼインが分解されるので、カゼインの分解に伴なって形成される菌体の周りのハローの大きさを指標として、親株であるバチルス・サブチリスDY−16株よりもプロテアーゼ生産能が低下した40株を分離した。
【0043】さらに、これらの株をLB培地で液体培養し、その培養上清に存在するプロテアーゼ活性を、FITC−カゼイン(ジクマ社)を基質として測定した。すなわち、バチルス・ズブチリスDY−16及びプロテアーゼ活性が低下した形質転換株40株をそれぞれ、LB培地10mlを用いて一晩培養し、その培養液1mlを遠心分離し、その上清をプロテアーゼ酵素液とした。
【0044】酵素液50μlと、0.2μgのFITC−カゼインとを、10mMのトリス−塩酸(pH7.5)および2mMのCaCl2 の緩衝液中(200μl)で37℃で2時間反応した。反応液に200μlの7.5%トリクロロ酢酸を添加し、遠心分離した後、その上清100μlに、500mMトリス−塩酸(PH8.5)900μlを添加し、プロテアーゼ活性を、螢光光度計を用い、波長600nmでの光学濃度(OD600 )により測定した。
【0045】なお、プロテアーゼ活性(PUD)は、37℃、pH7.5において、カゼインから1分間に1μモルのチロシンTyrに相当するトリクロロ酢酸可溶性ペプチドを遊離する酵素量を1PUDとして、算出した。その結果、プロテアーゼ活性が親株バチルス・サブチリスDY−16株に比べ極端に低下した4株(バチルス・サブチリスSPL9,11,14,39)を得た。
【0046】これら4株のプロテアーゼ活性をさらに詳しく測定した。バチルス・サブチリスDY−16株及びプロテアーゼ活性が低下した形質転換株4株を10mlのLB培地で18時間培養した後、50mlのMedium A培地に菌体を移し、培養24時間及び48時間後の培養上清中のプロテアーゼ活性を測定した。その結果を表2に示す。
【0047】
【表2】
得られたこれらの株は、親株に比べプロテアーゼ活性が1/20以下と低いことが確認された。特にバチルス・サブチリスSPL14株は、プロテアーゼ活性が低い。また、いずれの株においてもヒスチジンとロイシンに対する要求性を有しており、遺伝子組換え実験における宿主として要求される複数の栄養要求性を確認した。これらの菌株は、ペプチドを効率よく生産できる宿主として、安全かつ有効に用いることができる。
(2)α−アミラーゼ遺伝子のプローモーターの合成以下のような枯草菌α−アミラーゼ遺伝子のプローモーター配列をそれぞれ化学合成し、両遺伝子をアニーリングした後、pUC118のEcoRI−XbaIの間に挿入し、図2に示すようにプラスミドpMD3003を構築した。
【0048】
5'- AATTCAGGATCCTTGATTTGTATTCACTCTGCCAAGTTGTTTTGATAGAGTGATTGTGATAATTTAAAATGTAAGCGTGAACAAAATTCTCCAGT-3'5'-ACTGGAGAATTTTGTTCACGCTTACATTTTAAATTATCACAATCACTCTATCAAAACAACTTGGCAGAGTGAATACAAATCAAGATCCTG-3'(3)SD配列の合成枯草菌のα−アミラーゼ遺伝子及び黄色ブドウ球菌由来のプロテインA遺伝子のSD配列、及び枯草菌16SリボソームRNA(rRNA)の配列を参考として、以下の塩基配列で表されるSD配列を、ABI社DNA合成装置を用いて合成した。なお、遺伝子の5′側にはプローモーターとの連結部位であるXbaI部位を、3′領域にはα−アミラーゼ遺伝子のシグナル配列との連結部位であるNruI部位を導入した。
【0049】CTAGAAAGGAGGTGATCTAGAATTCATGTTCGTTTCCTCCACTACATCTTAAGTACAAGC(4)α−アミラーゼ遺伝子バチルス・サブチリス1A412株[オハイオ州立大学のバチルス・ジェネティク・ストック・センター(Bacillus Genetic Stock Center)(BGSC)より入手した]から前記斉藤・三浦の方法により調製した染色体DNA10μgを、100μlのSau3AI溶液[10mMのトリス−塩酸(pH7.5)、7mMのMgCl2 、100mMのNaCl)に溶解し、制限酵素Sau3AIを1単位加え、37℃で10分間反応させ、フェノール溶液[フェノールをTE緩衝液(10mMのトリス−塩酸(pH8.0)、1mMのEDTA)で飽和したもの]を添加し、反応を停止した後、エタノール沈澱物として、枯草菌染色体DNAのSau3AIの部分分解物を得た。
【0050】一方、1μgの枯草菌ベクターpUB110を、BamHI溶液[10mMのトリス−塩酸(pH8.0)、7mMのMgCl2 、100mMのNaCl、2mMの2−メルカプトエタノール、0.01%の牛血清アルブミン(BSA)]中で5単位の制限酵素BamHIと37℃で1時間反応させ、フェノール溶液で処理した後、エタノール沈澱物をpUB110のBamHI分解物として得た。
【0051】次いで、得られた上記二つのDNA断片のリゲーションを行った。すなわち、上記のようにして調製したそれぞれのDNA断片を、10μlのリゲーション溶液[66mMのトリス−塩酸(pH7.6)、6.6mMのMgCl2 、10mMのジチオスレイトール(DTT)、および0.1mMのアデノシン三リン酸(ATP)]に溶解した後、混合し、T4 DNAリガーゼ10単位を添加し、20℃で1時間反応を行った。
【0052】このDNA溶液を用いて、バチルス・サブチリス1A289株(BGSCより入手)をプロトプラスト化法(Chan, S., Cohen, N., Molec, gen. Genet., 168, 111-115 (1979) )により形質転換した。
【0053】形質転換株を寒天培地で培養し、カナマイシン耐性を示す約2万個のコロニーが得られ、寒天平板培地上でのヨード・デンプン反応により、このうち1株が菌体外にα−アミラーゼを分泌生産していることが確認された。
【0054】この株からプラスミドDNAを分離し、制限酵素による解析を行なったところ、図3に示すような制限酵素地図が得られ、前記プラスミドをpDCA100と命名した。pDCA100は、pUB110のBamHI部位に3.1KbのSau3AI断片が挿入された7.6Kbのプラスミドである。前記3.1KbのSau3AI断片の中には、枯草菌由来のα−アミラーゼ遺伝子[Yamazaki, H.et al., J. Bacteriol., 156 , 327 (1984)]と全く同じ制限酵素パターンが認められたことから、α−アミラーゼ遺伝子がクローニングされていることが確認された。
【0055】pDCA100のα−アミラーゼ遺伝子と同一と考えられるα−アミラーゼ遺伝子が導入された菌株は、アメリカ合衆国オハイオ大学バチルス・ジェネテック・ストック・センター(BGSC)にECE35株として登録されている。
(5)α−アミラーゼ遺伝子とSD配列の連結クローニングされたα−アミラーゼ遺伝子と合成SD配列とを、以下の塩基配列で表される合成遺伝子を介して連結した。
【0056】
5'-CGAAACGATTCAAAACCTCTTTACTGCCGTTATTTGCTGGATTTTTATTGCTGTTTTATTTGGTTCTGGCAGGAC-3'5'-CGGTCCTGCCAGAACCAAATAAAAACAGCAATAAAAATCCAGCGAATAACGGCAGTAAAGAGGTTTTGAATCGTTTTCG-3'この合成遺伝子は、α−アミラーゼ遺伝子のシグナル配列部分のアミノ酸配列(Yamazaki, H. et al., J. Bacteriol., 156 , 327 (1984))に従って塩基配列を決定し、化学合成した。常法に従って精製した後、アニーリングし、合成DNAリンカーとした。
(6)ターミネーターの作製枯草菌のアルカリプロテアーゼ遺伝子のターミネーターを参考として、以下のターミネーターをそれぞれ合成した。なお、5′末端側には、BglII部位を、3′末端側にはSphI部位を設けた。また、3′末端のすぐ近くにはBamHI部位を設けた。合成した後、両合成遺伝子をアニーリングし、ターミネーター断片とした。
【0057】
5'-GATCTATAAAAAGAAGCAGGTTCCTCCATACCTGCTTCTTTTTATTTGTCAGCATCCTGATGTTGGATCCGCATG-3'5'-CGGATCCAACATCAGGATGCTGACAAATAAAAAGAAGCAGGTATGGTGGAACCTGCTTCTTTTTATA-3'(7)SD配列、α−アミラーゼ遺伝子、ターミネーターの連結SD配列、α−アミラーゼ遺伝子、ターミネーターを、図4に示すように連結し、プラスミドpMD3062を得た。すなわち、DNA合成したSD配列、リンカーDNA、ターミネーター断片をDNAキナーゼを用いてカイネーションした。α−アミラーゼ遺伝子は、pDCA100から、HpaII−EcoRI断片、EcoRI−BclI断片として回収した。これらの遺伝子と、アルカリホスファターゼ処理したpUC19のXbaI−SphI切断断片と、α−アミラーゼ遺伝子部分を混合し、DNAリガーゼを用いて連結し、大腸菌HB101株を形質転換した。得られたアンピシリン耐性形質転換株の中から、目的とするプラスミドpMD3062を得た。
(8)pMD3082B1の構築とα−アミラーゼの生産プラスミドpMD3082B1の構築図を図5に示す。
【0058】先ず、pMD3003を制限酵素BamHIとXbaIとで切断し、プローモーターを切り出した。また、pMD3062をXbaIとBamHIとを用いて切断し、SD配列からターミネーターまでの領域を含むα−アミラーゼ遺伝子部分を得た。そして、これらの遺伝子を、アルカリホスファターゼ処理したpUB110のBamHI消化物と混合し、DNAリガーゼを用いて連結し、バチルス・サブチリスSPL14株をプロトプラスト化法により形質転換した。
【0059】得られたカナマイシン耐性形質転換株の中から、α−アミラーゼ生産能を示す200株の形質転換株を分離した。これらの形質転換株の中から、α−アミラーゼ遺伝子の方向性の異なる2種類のプラスミドが得られた。pUB110中のカナマイシン耐性遺伝子と同じ方向で遺伝子が導入されたプラスミドをpMD3082B1、その逆方向に導入されたプラスミドをpMD3082B2とした。それらの構成比は前者:後者=1:40であった。
【0060】次いで、これらのプラスミドを有するバチルス・サブチリスSPL14株を12時間浸透培養し、得られたα−アミラーゼ活性を比較検討したところ、pMD3082B1の方がpMD3082B2よりも約3倍のα−アミラーゼを生産し、培養12時間後には2000ユニット/mlのα−アミラーゼを生産した(表3参照)。なお、α−アミラーゼ活性は、不破らの方法(Fuwa, H., J. Biochem, 41, 583-603 (1954))に従って測定した。
【0061】
【表3】
(9)プローモーターを除去したときのα−アミラーゼ活性pMD3062をXbaIで切断し、SD配列、α−アミラーゼ遺伝子、ターミネーター遺伝子部分を回収し、制限酵素XbaIで切断したpUC118のXbaI切断部位に連結した。α−アミラーゼ遺伝子の5′側に、pUC118に存在するポリリンカー部位であるBamHI部位を位置させて連結したプラスミドをpMD30001とした。
【0062】このpMD30001を、制限酵素BamHIにより切断することにより、プローモーターが存在しないα−アミラーゼ遺伝子を回収できる。そこで、pMD3001をBamHIで切断した後、アルカリホスファターゼで処理したpUB110のBamHI切断断片と混合し、DNAリガーゼを用いて連結した後、バチルス・サブチリスSPL14株を形質転換した。得られたカナマイシン耐性形質転換株を培養し、それぞれプラスミドDNAを調製し、α−アミラーゼ遺伝子の方向性を調べたところ、pUB110にα−アミラーゼ遺伝子がカナマイシン耐性遺伝子と同じ方向に導入されたプラスミドpMD3082B1△と、逆方向に導入されたpMD3082B2△を得た。プラスミドpMD3082B1△およびpMD3082B2△の構築図を図6に示す。
【0063】それぞれのプラスミドを保有するバチルス・サブチリスSPL14株を培養し、得られた培養濾液中のα−アミラーゼ活性を調べたところ、表4に示す結果を得た。すなわち、pMD3082B2△を保有するバチルス・サブチリスSPL14株は、培養12時間後にα−アミラーゼを10ユニットしか生産せず、プラスミドを保有していないSPL14株の生産するα−アミラーゼ活性と変りがなかった。一方、pMD3082B1△を有するバチルス・サブチリスSPL14株は、培養12時間後にα−アミラーゼを800ユニット/ml生産した。
【0064】
【表4】
実施例2プラスミドpUB110を制限酵素SphIおよびEcoRIで切断し、SphI部位からEcoRI部位までの小さな約1Kbpの断片を得た。プラスミドpUC19を制限酵素SphIおよびEcoRIで切断し、pUC19のポリリンカーサイトに、DNAリガーゼを用いて前記約1Kbpの断片を導入し、プラスミドpUC19−110Bを構築した。プラスミドpUC19−110Bの構築図を図7に示す。
【0065】このプラスミドベクター上のpUB110部分に存在するBamHI部位を酵素BamHIにより解裂し、アルカリホスファターゼ処理した。また、前記pMD3082B1からα−アミラーゼ遺伝子部分を制限酵素BamHIで切断し回収した。両遺伝子断片を混合し、リガーゼにより連結し、大腸菌HB101株を形質転換した。
【0066】得られたアンピシリン耐性を有する形質転換株から、プラスミドを調製し、遺伝子の導入方向を調べたところ、供試48株のうち23株が、目的の方向に遺伝子が導入されたプラスミドを有していた。
【0067】得られたプラスミドを、制限酵素SphIとEcoRIとで切断し、DNA断片を調製した。また、pUB110を制限酵素SphIとEcoRIとで切断し、DNAリガーゼを用いて、前記DNA断片と連結することにより、遺伝子を、目的のカナマイシン耐性遺伝子と同じ方向に100%導入できた。
【図面の簡単な説明】
【図1】ベクタープラスミドpUB110の概略制限酵素地図である。
【図2】プラスミドpMD3003の構築図である。
【図3】プラスミドpDCA100の制限酵素地図である。
【図4】プラスミドpMD3062の構築図である。
【図5】プラスミドpMD3082B1の構築図である。
【図6】プラスミドpMD3082B1△及びpMD3082B2△の構築図である。
【図7】プラスミドpUC19−110Bの構築図である。
【特許請求の範囲】
【請求項1】 少なくともpUB110のSphI切断部位からBamHI切断部位までの塩基配列を含むpUB110由来のベクターであって、BamHI切断部位またはその近傍に、SphI切断部位からBamHI切断部位への方向に、SD(Shine-Dalgarno)配列を有するペプチド又は蛋白質をコードする遺伝子が連結されている高発現ベクター。
【請求項2】 ペプチド又は蛋白質をコードする遺伝子の3′側にターミネーターが連結されている請求項1記載の高発現ベクター。
【請求項3】 SD配列の5′側にプロモーターが連結されている請求項1又は2記載の高発現ベクター。
【請求項4】 請求項1〜3のいずれかに記載の高発現ベクターを保有するバチルス属細菌。
【請求項5】 請求項4記載のバチルス属細菌を培養し、産生したペプチド又は蛋白質を回収するペプチド又は蛋白質の製造方法。
【請求項1】 少なくともpUB110のSphI切断部位からBamHI切断部位までの塩基配列を含むpUB110由来のベクターであって、BamHI切断部位またはその近傍に、SphI切断部位からBamHI切断部位への方向に、SD(Shine-Dalgarno)配列を有するペプチド又は蛋白質をコードする遺伝子が連結されている高発現ベクター。
【請求項2】 ペプチド又は蛋白質をコードする遺伝子の3′側にターミネーターが連結されている請求項1記載の高発現ベクター。
【請求項3】 SD配列の5′側にプロモーターが連結されている請求項1又は2記載の高発現ベクター。
【請求項4】 請求項1〜3のいずれかに記載の高発現ベクターを保有するバチルス属細菌。
【請求項5】 請求項4記載のバチルス属細菌を培養し、産生したペプチド又は蛋白質を回収するペプチド又は蛋白質の製造方法。
【図1】

【図3】
【図4】
【図2】
【図5】
【図7】
【図6】
【図3】
【図4】
【図2】
【図5】
【図7】
【図6】
【公開番号】特開平5−252964
【公開日】平成5年(1993)10月5日
【国際特許分類】
【出願番号】特願平3−68988
【出願日】平成3年(1991)3月8日
【出願人】(000103079)エム・ディ・リサーチ株式会社 (2)
【公開日】平成5年(1993)10月5日
【国際特許分類】
【出願日】平成3年(1991)3月8日
【出願人】(000103079)エム・ディ・リサーチ株式会社 (2)
[ Back to top ]