
4’−C−置換−2−ハロアデノシン誘導体
【課題】優れた抗HIV作用を示す2−ハロアデノシン誘導体の製造方法の提供。
【解決手段】式(I)において5’位が水酸基、2位がアミノ基の化合物を出発物質として、水酸基保護工程、アミノ基のハロゲン基への変換工程、水酸基脱保護工程を経る式(I)の化合物の製造方法。

(式中、Xはフッ素原子又は塩素原子を示す)
【解決手段】式(I)において5’位が水酸基、2位がアミノ基の化合物を出発物質として、水酸基保護工程、アミノ基のハロゲン基への変換工程、水酸基脱保護工程を経る式(I)の化合物の製造方法。
(式中、Xはフッ素原子又は塩素原子を示す)
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、4’−C−置換−2−ハロアデノシン誘導体及びその医薬用途、特にエイズ(AIDS)の治療用用途に関するものである。
【背景技術】
【0002】
AZT(ジドブジン)、ddI(ジダノシン)、ddC(ザルシタビン)、d4T(スタブジン)、3TC(ラミブジン)などのヌクレオシド系逆転写酵素阻害剤(nucleoside reverse transcriptase inhibitors;NRTIs)にプロテアーゼ阻害剤(protease inhibitors;PIs)を加えた、いわゆる「強力な抗レトロウイルス剤による化学療法(highly active antiretroviral therapy;HAART)」と呼ばれる多剤併用療法によって、AIDSの臨床像は一変し、AIDSによる死亡者数も各国で激減した。
【0003】
しかし、HAARTによりAIDSによる死亡者が激減する一方で、複数の薬剤に対して交叉耐性を示す多剤耐性HIV−1(human immunodeficiency virus−1)株が出現し、たとえば、AZTと3TCの両方に耐性のHIVに感染した患者は、1990年初頭ではほとんど見られなかったのに対し、1995〜1996年になると42%にものぼっていることが報告されている。
【0004】
大類らは、2’−デオキシ−4’−C−エチニルヌクレオシドを合成し、それら化合物の抗HIV活性を測定した結果、特定の構造を有する2’−デオキシ−4’−C−エチニルヌクレオシドがAZTと同等あるいはAZTを凌ぐ優れた抗HIV活性を有すること、AZT、ddI、ddC、d4T、3TCなどの複数の抗HIV剤に耐性を有する多剤耐性ウイルス株にも有効なことを明らかにした(非特許文献1〜8、特許文献1〜3)。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】WO00/69876
【特許文献2】WO00/69877
【特許文献3】WO03/68796
【非特許文献】
【0006】
【非特許文献1】Nucleic Acids Symp Ser,Jan 2000;(44):105−6.
【非特許文献2】J Med Chem,Nov 2000;43(23):4516−25
【非特許文献3】Curr Drug Targets Infect Disord,May 2001;1(1):1−10
【非特許文献4】Antimicrob.Agents Chemother.,May 2001;45:1539−1546
【非特許文献5】Nucleosides Nucleotides Nucleic Acids,May 2003;22(5−8):887−9.
【非特許文献6】Chem.Pharm.Bull.,42(1994),p1688
【非特許文献7】J.Med.Chem.,39(1996),p3847
【非特許文献8】Bioorg Med Chem Lett,Nov 2003;13(21):3775−7
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明者らは、in vitroにおいて、種々の4’−C−置換ヌクレオシドの中でも特に優れた抗HIV活性を示した4’−C−エチニルプリンヌクレオシド誘導体及び4’−C−シアノプリンヌクレオシド誘導体について毒性の評価を行った。その結果、(1)最も強い抗HIV活性を示す2,6−ジアミノプリン誘導体及びグアニン誘導体はin vitro、in vivoにおいて毒性を示すこと、(2)毒性の低いアデニン誘導体は血中でアデノシンデアミナーゼにより速やかにヒポキサンチン誘導体へと変換され、抗HIV活性が減弱することが明らかとなった。
【課題を解決するための手段】
【0008】
本発明者らは、選択係数(細胞毒性濃度/抗HIV活性濃度)の更なる向上とアデノシンデアミナーゼによる不活性化に対する耐性付与を目的とし、種々の4’−C−置換プリンヌクレオシドの中で、強い抗HIV活性を示し、かつ毒性の低い4’−C−置換−2’−デオキシアデノシンをリード化合物とし、このリード化合物に化学的修飾を施すことにより数多くの誘導体を合成した。
【0009】
従来、アデノシンデアミナーゼによる不活性化に対する耐性付与に関しては、アデノシン誘導体の塩基部2位に電子吸引性のハロゲン原子を導入することにより、アデノシンデアミナーゼに対して若干の抵抗性を示すようになることが知られている(非特許文献6と7)が、ハロゲン原子の導入による選択係数の向上に関しては何ら示唆されていない。
【0010】
唯一、d4T(スタブジン;2’,3’−ジデヒドロ−3’−デオキシチミジン)の4’位にエチニル基を導入することで選択係数の向上が報告されているものの(非特許文献8)、基本骨格を著しく異にするプリン系ヌクレオシドであるアデノシン誘導体で同様の効果を期待することはできず、何ら参考になるものではなかった。
【0011】
新たに合成した誘導体の抗HIV活性等を検討した結果、リード化合物である2’−デオキシ−4’−C−エチニルアデノシンの塩基部2位にフッ素原子を導入した2’−デオキシ−4’−C−エチニル−2−フルオロアデノシンは、アデノシンデアミナーゼによる不活性化に対する耐性付与だけでなく、AZT、ddI、ddC、d4T、3TCなどの複数の抗HIV剤に耐性を有する多剤耐性ウイルス株にも有効で、しかも抗HIV活性が増強され、一方で、その細胞毒性は著しく減少することが明らかとなった。
【0012】
したがって、本発明は、この知見を更に発展させ、2−ハロアデニンを核酸塩基とし、糖部4位にエチニル基又はシアノ基を有する種々の4’−C−置換−2−ハロアデノシン誘導体を合成し、その活性を確認することにより完成されたものである。
【0013】
すなわち、本発明は、下記式[I]、[II]又は[III]で表される4’−C−置換−2−ハロアデノシン誘導体及び当該誘導体と薬学的に許容される担体とを含有してなる医薬組成物に関するものである。
【0014】
【化1】
【0015】
(式中、Xはハロゲン原子、R1はエチニル基又はシアノ基、R2は、水素原子、リン酸又はその誘導体残基を示す。)
また、本発明は、上記4’−C−置換−2−ハロアデノシン誘導体又はそれを含む医薬組成物をヒトを含む動物に投与することを特徴とするエイズの治療方法に関するものである。
【発明の効果】
【0016】
本発明化合物は、後述試験例に示すように、たとえば、2’−デオキシ−4’−C−エチニル−2−フルオロアデノシンは、アデノシンデアミナーゼによる不活性化に対する耐性付与だけでなく、AZT、ddI、ddC、d4T、3TCなどの複数の抗HIV剤に耐性を有する多剤耐性ウイルス株にも有効で、しかもリード化合物である2’−デオキシ−4’−C−エチニルアデノシンの抗HIV活性と比較して144倍と予想できないほどに抗HIV活性が増強され、一方で、その細胞毒性は著しく減少することが明らかとなった。この結果、この化合物の選択係数は110,000となり、従来の2’−デオキシ−4’−C−エチニルアデノシン(EdAdo)の1,630を遙かに上回る驚くべき結果となった。
このように、本発明化合物は、優れた抗HIV作用、特にAZT、DDI、DDC、D4T、3TCなどの抗HIV剤の複数の薬剤に耐性を有する多剤耐性HIV株にも有効で、細胞毒性も低く、アデノシンデアミナーゼによる不活性化に対する耐性も付与されていることから、医薬品、特にエイズ治療薬として有用である。
【図面の簡単な説明】
【0017】
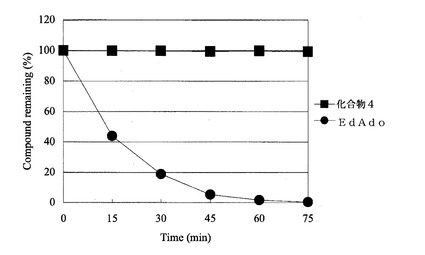
【図1】アデノシンデアミナーゼによる脱アミノ化反応における化合物の安定性を示したものである。黒四角は本発明化合物2’−デオキシ−4’−C−エチニル−2−フルオロアデノシンの結果を、黒丸は公知の2’−デオキシ−4’−C−エチニルアデノシンの結果を示したものである。
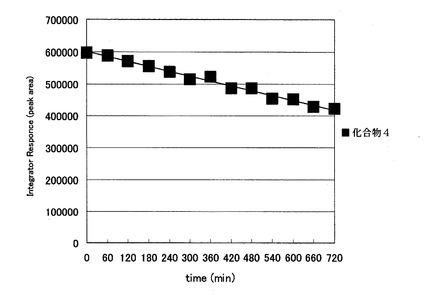
【図2】酸性条件下における本発明化合物2’−デオキシ−4’−C−エチニル−2−フルオロアデノシンの安定性を示したものである。
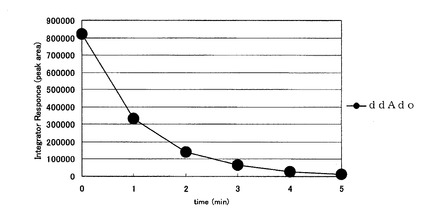
【図3】酸性条件下における公知の2’,3’−ジデオキシアデノシン(ddAdo)の安定性を示したものである。
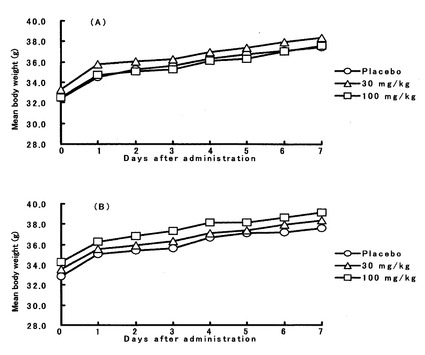
【図4】本発明化合物2’−デオキシ−4’−C−エチニル−2−フルオロアデノシンを投与後のマウスの体重変化を示したものである。図4中のグラフAは経口投与時の結果を、グラフBは静脈内投与時の結果を示す。なお、各グラフ中の丸印はプラセボを示し、三角印、四角印は投与量がそれぞれ30mg/kg、100mg/kgのときの結果を示す。
【発明を実施するための形態】
【0018】
(1)化合物
本発明化合物は、前記式[I]、[II]又は[III]で表されるものである。上記式中、R2のリン酸又はその誘導体残基としては、リン酸残基、ジリン酸残基、トリリン酸残基、ホスホン酸残基、及びこれらのジエステル、トリエステル等のポリエステル、モノアミデート、ジアミデート等のアミデート、チオエート、セレノエート、ボラノエート等を例示することができる。また、Xのハロゲン原子としては、臭素原子、ヨウ素原子、フッ素原子又は塩素原子を例示することができる。
【0019】
このような化合物のうち、(a)R2が水素原子又はホスホン酸残基、(b)Xがフッ素原子又は塩素原子、(c)R1がエチニル基である条件の1つ又は複数を満たすものを好適な化合物として例示することができる。具体的には、以下の化合物を挙げることができる。
【0020】
<式[I]化合物>
2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン、4’−C−シアノ−2’−デオキシ−2−フルオロアデノシン、2−クロロ−2’−デオキシ−4’−C−エチニルアデノシン又は2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン5’−H−ホスホネート
【0021】
<式[II]化合物>
2’,3’−ジデヒドロ−2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン、2’,3’−ジデヒドロ−2’,3’−ジデオキシ−4’−C−シアノ−2−フルオロアデノシン、2’,3’−ジデヒドロ−2’,3’−ジデオキシ−4’−C−エチニル−2−クロロアデノシン又は2’,3’−ジデヒドロ−2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン5’−H−ホスホネート
【0022】
<式[III]化合物>
2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン、2’,3’−ジデオキシ−4’−C−シアノ−2−フルオロアデノシン、2’,3’−ジデオキシ−4’−C−エチニル−2−クロロアデノシン又は2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン5’−H−ホスホネート
【0023】
本発明化合物は、塩、水和物又は溶媒和物の形態であってもよい。そのような塩としては、R2が水素原子である場合には塩酸塩又は硫酸塩などの酸付加物、R2がリン酸残基である場合にはナトリウム塩、カリウム塩又はリチウム塩などのアルカリ金属塩、カルシウム塩などのアルカリ土類金属塩もしくはアンモニウム塩などの薬学的に許容される任意の塩が例示される。
また、水和物又は溶媒和物としては、本発明の化合物又はその塩1分子に対し、0.1〜3.0分子の水又は溶媒が付着したものを例示することができる。更に、本発明の化合物には、互変異性体などの各種異性体も包含されうる。
【0024】
(2)製造法
本発明化合物[I]は、以下に説明する工程により製造することができる。
第1工程;
第1工程は式[IV]で表される化合物の3’位水酸基及び5’位水酸基を保護し、式[V]で表される化合物を得る工程である。
【0025】
【化2】
【0026】
(式中、Pは保護基,R1はエチニル基又はシアノ基を示す。)
【0027】
式[IV]で表される原料化合物のうち、R1がエチニル基である化合物はJ.Med.Chem.,43,4516−4525(2000)に記載されており、R1がシアノ基である化合物はWO03/68796に記載されており、公知である。
Pで表される3’,5’位水酸基の保護基としては、水酸基の保護基として常用されているものであればよく、たとえばエーテル系保護基、アシル系保護基、シリル系保護基、アセタール系保護基などを例示することができる。より具体的には、エーテル系保護基としては、メチルエーテル、第3級ブチルエーテル、ベンジルエーテル、メトキシベンジルエーテル、トリチルエーテルなどを、アシル系保護基としてはアセチル、ベンゾイル、ピバロイルなどを、シリル系保護基としてはt−ブチルジメチルシリル、t−ブチルジフェニルシリル、トリメチルシリル、トリエチルシリルなどを、アセタール系保護基としてはイソプロピリデン、エチリデン、メチリデン、ベンジリデン、テトラヒドロピラニル、メトキシメチルなどをそれぞれ使用することができる。
【0028】
保護基の導入は常法によって行うことができる。たとえば、ピリジン、アセトニトリル、ジメチルホルムアミド等の有機溶媒中、アルカリ金属アルコキシド、トリエチルアミン、4−ジメチルアミノピリジン、イミダゾール等の塩基の存在下、式(IV)の化合物と保護基導入試薬(例えば、アルキルハライド、酸ハライド、酸無水物、アルキルシリルハライド等)とを、−10〜100℃で反応させることによって実施することができる。
【0029】
第2工程;
第2工程は、式[V]で表される化合物の2位アミノ基をハロゲン原子へと変換し、式[VI]で表される化合物を得る工程である。
【0030】
【化3】
【0031】
(式中、Pは保護基、Xはハロゲン原子、R1はエチニル基又はシアノ基を示す。)
【0032】
式[VI]で表される化合物は、式[V]で表される化合物の2位アミノ基を亜硝酸誘導体で処理後、ハロゲン化試薬を用いて塩基部2位にハロゲン原子を導入するか、又は、2,6位のアミノ基を同条件下処理し、2,6−ジハロプリン誘導体とした後、アンモニア処理により塩基部6位のハロゲン原子をアミノ基とすることで合成することができる。
【0033】
式[V]で表される化合物の2位アミノ基をフッ素へと置換する試薬としては、テトラフルオロホウ酸中の亜硝酸ナトリウム、フッ化水素−ピリジン中の亜硝酸t−ブチル等の亜硝酸エステルなどを例示することができる。
【0034】
反応条件は用いる試薬により異なり、たとえば、フッ化水素−ピリジン中、亜硝酸t−ブチルを用いる場合、フッ化水素−ピリジンを溶媒として、亜硝酸t−ブチルを1〜3モル用い、−50℃から室温で15分から5時間程度反応させればよい。また、2,6−ジフルオロプリン誘導体とした場合は、引き続き、ジオキサン、メタノール等の有機溶媒中、アンモニア水処理すればよい。
【0035】
一方、式[V]で表される化合物の2位アミノ基を塩素へと置換する試薬としては、ジクロロメタンなどの有機溶媒中、三塩化アンチモン−亜硝酸t−ブチル、塩化アセチル−亜硝酸ベンジルトリエチルアンモニウムの組み合わせなどを例示することができる。
【0036】
反応条件は用いる試薬により異なり、たとえば、塩化アセチル−亜硝酸ベンジルトリエチルアンモニウムの組み合わせを用いる場合、ジクロロメタン等の有機溶媒中、化合物[V]1モルに対して、亜硝酸ベンジルトリエチルアンモニウム1〜5モルと1〜5モルの塩化アセチルとを−50℃から室温で30分間から3時間程度処理した後、化合物[V]を−50℃から室温で1時間から数日間反応させればよい。また、2,6−ジクロロプリン誘導体とした場合は、引き続き、ジオキサン、メタノール等の有機溶媒中、アンモニア水処理すればよい。
【0037】
こうして得られた化合物[VI]の保護基を除去して、R2が水素である本発明化合物を得、必要に応じてリン酸化して本発明化合物を得る。
【0038】
【化4】
【0039】
(式中、Pは保護基、Xはハロゲン原子、R1はエチニル基又はシアノ基、R2は水素原子、リン酸又はその誘導体残基を示す。)
保護基の除去は、使用した保護基に応じ、酸性加水分解、アルカリ性加水分解、フッ化テトラブチルアンモニウム処理、接触還元などの通常の処理方法から適宜選択して行えばよい。
【0040】
【化5】
【0041】
(式中、Xはハロゲン原子、R1はエチニル基又はシアノ基、R2は水素を示す。)
【0042】
本発明化合物の5’−H−ホスホネート誘導体[VII]を得るには、有機溶媒中、適当な縮合剤を用い、R2が水素である化合物[I]とホスホン酸を縮合すればよい。用いる有機溶媒として、ピリジン、又はトリエチルアミン等の塩基存在下でのジメチルホルムアミド、縮合剤としては、ジシクロヘキシルカルボジイミド、ジイソプロピルカルボジイミド、水溶性カルボジイミド等のカルボジイミド、塩化トルエンスルホニル等のスルホン酸ハロゲン化物、塩化ジフェニルリン酸等のリン酸塩化物を例示することができる。
【0043】
反応条件は用いる試薬により異なり、たとえば、ピリジン中、ジシクロヘキシルカルボジイミドを用いる場合、化合物[I]1モルに対して、ホスホン酸を1〜5モル、ジシクロヘキシルカルボジイミドを1〜10モル用い、0℃〜50℃で1〜24時間程度反応させることにより実施できる。
【0044】
また、R2がモノリン酸残基である化合物を得る場合には、R2が水素原子である化合物をオキシ塩化リン、テトラクロロピロリン酸などのヌクレオシドの5’位の選択的なリン酸化に使用されるリン酸化剤と反応させることにより、更に、R2がジリン酸、トリリン酸残基である化合物を得るには、対応する5’−モノリン酸化合物をホスホイミダゾリド、ホスホモルホリデート、ジフェニルリン酸無水物などを用いて活性化した後、それぞれ、リン酸、ピロリン酸、又はこれらの適当な塩と反応させることにより、遊離酸型又は塩型の目的化合物を得ることができる。
【0045】
本発明化合物[II]は、以下に説明する工程により製造することができる。
第1工程;
第1工程は式[I]で表され、R2が水素である化合物の5’位水酸基を選択的に保護し、式[VIII]で表される化合物を得る工程である。
【0046】
【化6】
【0047】
(式中、Pは保護基、Xはハロゲン原子、R1はエチニル基又はシアノ基、R2は水素を示す。)
【0048】
Pで表される5’位水酸基の保護基としては、1級水酸基の選択的な保護基として常用されているものであればよく、具体的には、トリメトキシトリチル基、ジメトキシトリチル基、メトキシトリチル基、トリチル基、t−ブチルジメチルシリル基、t−ブチルジフェニルシリル基、ベンゾイル基等を例示することができる。
【0049】
保護基の導入反応は、前記と同様の方法にて実施できる。
【0050】
第2工程;
第2工程は、式[VIII]で表される化合物の3’位水酸基を脱水し、2’,3’−2重結合とし、化合物式[VIV]とする工程である。
【0051】
【化7】
【0052】
(式中、Pは保護基、Xはハロゲン原子、R1はエチニル基又はシアノ基を示す。)
化合物[VIII]の3’位水酸基を脱水し、化合物[VIV]とするには、化合物[VIII]の3’位水酸基をハロゲン原子、又は、メタンスルホン酸、クロロメタンスルホン酸、トルエンスルホン酸、トリフルオロメタンスルホン酸等のスルホン酸エステルのような脱離能を有する官能基へと変換後、これを塩基処理により脱離させることで実施できる。
【0053】
反応条件は用いる試薬により異なり、たとえば、トリフルオロメタンスルホン酸エステルを経る反応を用いる場合、ジクロロメタン、ピリジン等の有機溶媒中、トリフルオロメタンスルホン酸無水物とピリジン、トリエチルアミン等の塩基をそれぞれ、1〜5モル、5〜10モル用い、−78℃〜室温で1時間〜24時間程度反応させることにより実施できる。
【0054】
こうして得られた化合物[VIV]の保護基を除去して、R2が水素である本発明化合
物を得、必要に応じてリン酸化して本発明化合物を得る。
【0055】
【化8】
【0056】
(式中、Xはハロゲン原子、Pは保護基、R1はエチニル基又はシアノ基、R2は、水素原子、リン酸又はその誘導体残基を示す。)
【0057】
保護基の除去は、使用した保護基に応じ、酸性加水分解、アルカリ性加水分解、フッ化テトラブチルアンモニウム処理、接触還元などの通常の処理方法から適宜選択して行えばよい。
【0058】
R2がリン酸又はその誘導体残基を示す化合物は、前記式[I]化合物と同様の方法で合成することができる。
【0059】
本発明化合物[III]は、以下に説明する工程により製造することができる。
第1工程;
第1工程は、式[X]で表される化合物の4位のヒドロキシメチル基を酸化してアルデヒド化合物とし、更にトリエチルシリルエチニル化合物、又はシアノ化合物へと変換することにより式[XI]で表される化合物を得る工程である。
【0060】
【化9】
【0061】
(R1はエチニル基、トリエチルシリルエチニル基又はシアノ基を示す。)
【0062】
原料化合物は、式[X]で表される公知化合物である(Biosci.Biotech.Biochem.,57,1433−1438(1993))。トリエチルシリルエチニル化合物への変換反応は、式[X]化合物の4位ヒドロキシメチル基をホルミル基へと酸化した後、これをジブロモビニル基へと変換し、引き続き、強塩基処理するにより脱ブロモ化水素することにより実施できる。
【0063】
式[X]で表される化合物の4−ヒドロキシメチル基をホルミル基に変換する場合、用いる酸化剤としては、無水クロム酸、ピリジンと無水酢酸との複合試薬、ピリジンクロロクロメート、ピリジンジクロメートなどのクロム系酸化剤;デス−マーチン試薬などの高原子価ヨウ素酸化剤;ジメチルスルホキシドと無水酢酸、塩化オキサリル又はジシクロヘキシルカルボジイミドとを組み合わせて用いるジメチルスルホキシド系酸化剤などを例示することができる。
【0064】
反応条件は用いる酸化剤により異なり、たとえば、塩化オキサリルとジメチルスルホキシドを用いて酸化する場合、ジクロロメタンなどの有機溶媒中、必要によりアルゴン、窒素などの不活性ガス雰囲気下、式[X]化合物1モルに対して塩化オキサリルとジメチルスルホキシドをそれぞれ1〜5モル、1.5〜6モル用い、−100〜0℃で15分から2時間程度反応させ、トリエチルアミンなどの塩基類を2〜10モル添加し、更に室温にて15分から2時間程度反応させることにより実施できる。
【0065】
次に、得られたアルデヒド化合物からアルキン化合物への変換は、アルデヒド化合物を増炭反応(C−C結合形成反応)に付し、強塩基で処理してメタルアルキニル化合物とし、最後に保護基を導入することにより実施できる。増炭反応は、ジクロロメタン、ジクロロエタンなどの有機溶媒中、必要によりアルゴン、窒素などの不活性ガス雰囲気下、先に得られたアルデヒド化合物1モルに対して四臭化炭素とトリフェニルホスフィンをそれぞれ1〜5モルと2〜10モル用い、0〜50℃で15分〜3時間程度反応させることにより実施できる。
【0066】
強塩基処理は、テトラヒドロフラン、1,4−ジオキサン、ジメトキシエタンなどの有機溶媒中、必要によりアルゴン、窒素などの不活性ガス雰囲気下、増炭反応により得られた化合物1モルに対してメチルリチウム、n−ブチルリチウム、t−ブチルリチウムなどのリチウム化合物2〜4モル用い、−100〜−20℃で5〜60分程度反応させることにより実施できる。更に、得られた化合物のアルキニル基にシリル系保護基を導入する場合、上記強塩基処理に続けてクロロトリエチルシランなどのシリル化剤を添加し、反応させることにより実施することができる。
【0067】
一方、式[X]化合物のシアノ化合物への変換反応は、式[X]化合物の4位ヒドロキシメチル基をホルミル基へと酸化した後、これをオキシム化合物へと変換し、引き続き、得られたオキシム化合物を脱水することにより実施できる。
【0068】
式[X]で表される化合物の4−ヒドロキシメチル基をホルミル基に変換する場合、用いる酸化剤としては、無水クロム酸、ピリジンと無水酢酸との複合試薬、ピリジンクロロクロメート、ピリジンジクロメートなどのクロム系酸化剤;デス−マーチン試薬などの高原子価ヨウ素酸化剤;ジメチルスルホキシドと無水酢酸、塩化オキサリル又はジシクロヘキシルカルボジイミドとを組み合わせて用いるジメチルスルホキシド系酸化剤などを例示することができる。
【0069】
反応条件は用いる酸化剤により異なり、たとえば、塩化オキサリルとジメチルスルホキシドを用いて酸化する場合、ジクロロメタンなどの有機溶媒中、必要によりアルゴン、窒素などの不活性ガス雰囲気下、式[X]化合物1モルに対して塩化オキサリルとジメチルスルホキシドをそれぞれ1〜5モル、1.5〜6モル用い、−100〜0℃で15分から2時間程度反応させ、トリエチルアミンなどの塩基類を2〜10モル添加し、更に室温にて15分から2時間程度反応させることにより実施できる。
【0070】
アルデヒド化合物のオキシム化合物への変換は、ピリジン等の有機溶媒中、アルデヒド化合物1モルに対して1〜5モルの塩酸ヒドロキシルアミンを使用し、室温〜100℃で30分〜3時間程度反応させることにより実施できる。
【0071】
オキシム化合物の脱水は、ジクロロメタン、アセトニトリル、テトラヒドロフラン等の有機溶媒中、ピリジン、トリエチルアミン、酢酸ナトリウム等の塩基存在下、ホスゲン、カルボニルジイミダゾール、塩化メタンスルホニル、無水酢酸等の脱水剤を用いることで実施できる。
【0072】
脱水反応条件は用いる脱水剤により異なり、たとえば、塩化メタンスルホニルを用いて脱水する場合、ジクロロメタン、テトラヒドロフラン、ピリジン等の有機溶媒中、オキシム化合物1モルに対して塩化メタンスルホニルとトリエチルアミンをそれぞれ1〜5モル、5〜10モル用い、−50℃〜室温で15分〜2時間程度反応させることにより実施できる。
【0073】
第2工程;
第2工程は、式[XI]で表される化合物の3,5位の水酸基を保護するメトキシベンジリデン基を除去し、式[XII]で表される化合物とする工程である。
【0074】
【化10】
【0075】
(R1はエチニル基、トリエチルシリルエチニル基又はシアノ基を示す。)
【0076】
保護基の除去法としては、酸性加水分解、接触還元等の処理方法から適宜選択して行えばよい。
【0077】
反応条件は用いる反応により異なり、たとえば、酸性加水分解を用いるときは、式[XI]の化合物をギ酸、酢酸等の有機酸、又は鉱酸の水溶液中、0〜100℃で1〜24時間反応させることにより実施できる。
【0078】
第3工程;
第3工程は式[XII]で表される化合物の5位水酸基を選択的に保護し、式[XIII]で表される化合物を得る工程である。
【0079】
【化11】
【0080】
(式中、Pは保護基、R1はエチニル基、トリエチルシリルエチニル基又はシアノ基を示す。)
【0081】
Pで表される5位水酸基の保護基としては、1級水酸基の選択的な保護基として常用されているものであればよく、具体的には、トリメトキシトリチル基、ジメトキシトリチル基、メトキシトリチル基、トリチル基、t−ブチルジメチルシリル基、t−ブチルジフェニルシリル基、ベンゾイル基等を例示することができる。
保護基の導入反応は、前記と同様の方法にて実施できる。
【0082】
第4工程;
第4工程は式[XIII]で表される化合物の3位水酸基を還元し、式[XIV]で表される化合物を得る工程である。
【0083】
【化12】
【0084】
(式中、Pは保護基、R1はエチニル基、トリエチルシリルエチニル基又はシアノ基を示す。)
【0085】
3位水酸基のデオキシ化は、3位水酸基をハロゲン化体(ヨウ素体、臭素体、塩素体)、フェノキシチオカルボニル体、チオカルボニルイミダゾール体、メチルジチオカルボネート体等に変換した後、ラジカル開始剤存在下、ラジカル還元剤により還元することによって行うことができる。
【0086】
例えば、フェノキシチオカルボニル体に導いてデオキシ化する場合、フェノキシチオカルボニル化反応は、必要によりアルゴン、窒素等の不活性ガス雰囲気下、テトラヒドロフラン、アセトニトリル、ジクロロメタン等の有機溶媒中、ジメチルアミノピリジン、ピリジン等の塩基共存下、3位水酸基の保護基のみ除去された上記化合物1モルに対してクロロチオノギ酸フェニル誘導体1〜10モル、好ましくは1〜2モル用い、0〜50℃で0.5〜5時間程度撹拌反応させることにより実施することができる。
【0087】
続けて行う還元反応は、トルエン、ベンゼン等の有機溶媒中、必要によりアルゴン、窒素等の不活性ガス雰囲気下、アゾビスイソブチロニトリル等のラジカル開始剤存在下、上記フェノキシチオカルボニル体1モルに対して水素化トリブチルスズ、トリス(トリメチルシリル)シラン等のラジカル還元剤1〜10モル、好ましくは2〜5モル用い、50〜150℃で1〜5時間程度撹拌反応させることにより実施することができる。
【0088】
第5工程;
第5工程は式[XIV]で表される化合物の1,2位イソプロピリデン基を除去後、生じた水酸基をアセチル化し、式[XV]で表される化合物を得る工程である。
【0089】
【化13】
【0090】
(式中、Pは保護基、R1はエチニル基、トリエチルシリルエチニル基又はシアノ基を示す。)
【0091】
1,2位イソプロピリデン基の除去法として、酸性加水分解の条件を用いるときは、式[XIV]化合物をギ酸、酢酸等の有機酸、又は鉱酸の水溶液中、0〜100℃で1〜24時間反応させることにより実施できる。
【0092】
引き続く、水酸基へのアセチル基の導入は、常法により行うことができ、たとえば、ピリジン、アセトニトリル、ジクロロメタン等の有機溶媒中、ピリジン、トリエチルアミン等の塩基存在下、塩化アセチル、無水酢酸などのアセチル化剤と反応させることにより実施することができる。
【0093】
たとえば、ピリジン中、無水酢酸により反応を行う場合、上記の脱イソプロピリデン体1モルに対して、2〜10モルの無水酢酸、及び、場合に応じて、触媒量の4−ジメチルアミノピリジンを用い、0〜100℃で1〜24時間反応させることにより実施できる。
【0094】
第6工程;
第6工程は式[XV]で表される化合物を、2,6−ジアミノプリンと縮合し、式[XVI]で表される化合物を得る工程である。
【0095】
【化14】
【0096】
(式中、Pは保護基、R1はエチニル基、トリエチルシリルエチニル基又はシアノ基を示す。)
【0097】
式[XV]で表される化合物と2,6−ジアミノプリンとの縮合は、ルイス酸存在下、式[XV]の化合物を2,6−ジアミノプリンと反応させることによって行うことができる。2,6−ジアミノプリンはシリル化したものを用いてもよく、このようなシリル化した塩基類は公知の方法、たとえばヘキサメチルジシラザンとトリメチルクロロシラン中で加熱環流するか、又はアセトニトリル、1,2−ジクロロエタン等の有機溶媒中、ビス(トリメチルシリル)アセトアミドと加熱還流することにより得ることができる。使用するルイス酸としては、トリフルオロメタンスルホン酸トリメチルシリル、四塩化すず、塩化亜鉛、ヨウ化亜鉛、無水塩化アルミニウムなどが例示される。
【0098】
縮合反応は、ジクロロメタン、1,2−ジクロロエタン、アセトニトリル、トルエン等の有機溶媒中、必要によりアルゴン、窒素などの不活性ガス雰囲気下、式[XV]の化合物1モルに対し2,6−ジアミノプリン1〜10モル及びルイス酸0.1〜10モルとを用い、−20〜150℃で30分〜24時間程度反応させることにより実施することができる。
【0099】
第7工程;
第7工程は、式[XVI]で表される化合物の2位アミノ基をフッ素又は塩素へと変換し、式[XVII]で表される化合物を得る工程である。
【0100】
【化15】
【0101】
(式中、Pは保護基、Xはハロゲン原子、R1はエチニル基、トリエチルシリルエチニル基又はシアノ基を示す。)
【0102】
式[XVII]で表される化合物は、式[XVI]で表される化合物の2位アミノ基を亜硝酸誘導体で処理後、ハロゲン化試薬を用いて塩基部2位にハロゲン原子を導入するか、又は、2,6位のアミノ基を同条件下処理し、2,6−ジハロプリン誘導体とした後、アンモニア処理により塩基部6位のハロゲン原子をアミノ基とすることで合成することができる。
【0103】
式[XVI]で表される化合物の2位アミノ基をフッ素へと置換する試薬としては、テトラフルオロホウ酸中の亜硝酸ナトリウム、フッ化水素−ピリジン中の亜硝酸t−ブチル等の亜硝酸エステルなどを例示することができる。
【0104】
反応条件は用いる試薬により異なり、たとえば、フッ化水素−ピリジン中、亜硝酸t−ブチルを用いる場合、フッ化水素−ピリジンを溶媒として、亜硝酸t−ブチルを1〜3モル用い、−50℃から0℃で15分から5時間程度反応させればよい。また、2,6−ジフルオロプリン誘導体とした場合は、引き続き、ジオキサン、メタノール等の有機溶媒中、アンモニア水処理すればよい。
【0105】
一方、式[XVI]で表される化合物の2位アミノ基を塩素へと置換する試薬としては、ジクロロメタンなどの有機溶媒中、三塩化アンチモン−亜硝酸t−ブチル、塩化アセチル−亜硝酸ベンジルトリエチルアンモニウムの組み合わせなどを例示することができる。
【0106】
反応条件は用いる試薬により異なり、たとえば、塩化アセチル−亜硝酸ベンジルトリエチルアンモニウムの組み合わせを用いる場合、ジクロロメタン等の有機溶媒中、化合物[XVI]1モルに対して、亜硝酸ベンジルトリエチルアンモニウム1〜5モルを−50℃から室温で30分間から3時間程度、1〜5モルの塩化アセチルと処理した後、化合物[XVI]を−50℃から室温で1時間から数日間反応させればよい。また、2,6−ジクロロプリン誘導体とした場合は、引き続き、ジオキサン、メタノール等の有機溶媒中、アンモニア水処理すればよい。
【0107】
第8工程;
第8工程は、式[XVII]で表される化合物の2’位水酸基を保護するアセチル基を除去し、式[XVIII]で表される化合物を得る工程である。
【0108】
【化16】
【0109】
(式中、Pは保護基、Xはハロゲン原子、R1はエチニル基、トリエチルシリルエチニル基又はシアノ基を示す。)
【0110】
アセチル基の除去は、適当な塩基又は酸触媒を用いて行うことができ、例えば塩基触媒を用いる場合、エタノール等のアルコール系溶媒と水との混合溶媒中、水酸化ナトリウム、水酸化カリウム、トリエチルアミン、アンモニア水等を例示することができる。
【0111】
例えば、メタノール中、アンモニア水を用い、0〜100℃で1〜24時間反応させることにより、実施できる。
【0112】
第9工程;
第9工程は、式[XVIII]で表される化合物の2’位水酸基を還元し、式[XIX]で表される化合物を得る工程である。
【0113】
【化17】
【0114】
(式中、Pは保護基、Xはハロゲン原子、R1はエチニル基、トリエチルシリルエチニル基又はシアノ基を示す。)
【0115】
2’位水酸基のデオキシ化は、2’位水酸基をハロゲン化体(ヨウ素体、臭素体、塩素体)、フェノキシチオカルボニル体、チオカルボニルイミダゾール体、メチルジチオカルボネート体等に変換した後、ラジカル開始剤存在下、ラジカル還元剤により還元することによって行うことができる。
【0116】
例えば、フェノキシチオカルボニル体に導いてデオキシ化する場合、フェノキシチオカルボニル化反応は、必要によりアルゴン、窒素等の不活性ガス雰囲気下、テトラヒドロフラン、アセトニトリル、ジクロロメタン等の有機溶媒中、ジメチルアミノピリジン、ピリジン等の塩基共存下、2’位水酸基の保護基のみ除去された上記化合物1モルに対してクロロチオノギ酸フェニル誘導体1〜10モル、好ましくは1〜2モル用い、0〜50℃で0.5〜5時間程度撹拌反応させることにより実施することができる。
【0117】
続けて行う還元反応は、トルエン、ベンゼン等の有機溶媒中、必要によりアルゴン、窒素等の不活性ガス雰囲気下、アゾビスイソブチロニトリル等のラジカル開始剤存在下、上記フェノキシチオカルボニル体1モルに対して水素化トリブチルスズ、トリス(トリメチルシリル)シラン等のラジカル還元剤1〜10モル、好ましくは2〜5モル用い、50〜150℃で1〜5時間程度撹拌反応させることにより実施することができる。
【0118】
こうして得られた化合物[XIX]の水酸基、及び、エチニル基の保護基を除去して、R2が水素である本発明化合物を得、必要に応じてリン酸化して本発明化合物を得る。
【0119】
【化18】
【0120】
(式中、Pは保護基、Xはハロゲン原子、R1はエチニル基、トリエチルシリルエチニル基又はシアノ基、R2は、水素、リン酸又はその誘導体残基を示す。)
【0121】
保護基の除去は、使用した保護基に応じ、酸性加水分解、アルカリ性加水分解、フッ化テトラブチルアンモニウム処理、接触還元などの通常の処理方法から適宜選択して行えばよい。
また、R2がリン酸又はその誘導体残基を示す化合物は、前記式[I]化合物と同様の方法で合成することができる。
【0122】
本発明化合物は、一般のヌクレオシド、ヌクレオチドの単離精製に使用されている方法(例えば、再結晶法、イオン交換カラムクロマトグラフィー、吸着カラムクロマトグラフィーなど)を適宜組み合せて分離精製することができる。このようにして得られた化合物は、必要に応じて塩とすることもできる。
【0123】
(3)用途
本発明化合物は、後述の試験例に示すようにレトロウイルスに対して優れた抗ウイルス作用を有することから、これらを有効成分とする本発明組成物は医薬としての使用、具体的にはレトロウイルスの感染症の処置、特にヒト免疫不全ウイルス(HIV)感染に起因するエイズの治療に有用である。
【0124】
本発明化合物の投与量は、患者の年齢、体重、疾病、患者の重篤度、薬物による忍容性、投与方法などにより異なり、これらの条件を総合した上で適宜決定されるものであるが、通常1日当たり0.00001〜1000mg/kg体重、好ましくは0.0001〜100mg/kg体重の範囲内から選ばれ、一回又は複数回に分けて投与される。
投与方法は、経口、非経口、経腸、局所投与などのいずれの経路でもよい。
【0125】
本発明化合物の製剤化に際しては、通常使用される製剤用担体、賦形剤、その他の添加剤を含む組成物として使用するのが普通である。担体としては、乳糖、カオリン、ショ糖、結晶セルロース、コーンスターチ、タルク、寒天、ペクチン、ステアリン酸、ステアリン酸マグネシウム、レシチン、塩化ナトリウムなどの固体状担体、グリセリン、落花生油、ポリビニルピロリドン、オリーブ油、エタノール、ベンジルアルコール、プロピレングリコール、水などの液状担体を例示することができる。
【0126】
剤型としては任意の形態を採ることができ、たとえば固体状担体を使用する場合には錠剤、散剤、顆粒剤、カプセル化剤、坐剤、トローチ剤などを、液状担体を使用する場合にはシロップ、乳液、軟ゼラチンカプセル、クリーム、ゲル、ペースト、スプレー、注射などをそれぞれ例示することができる。
【実施例】
【0127】
以下、本発明を合成例、試験例、製剤例などをあげて具体的に説明するが、本発明はこれらによって何等限定されるものではない。
【0128】
合成例1:2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン(化合物4)の合成
【0129】
(1)9−(3,5−ジ−O−アセチル−2−デオキシ−4−C−エチニル−β−D−リボ−ペントフラノシル)−2,6−ジアミノプリン(化合物2)の合成
【0130】
【化19】
【0131】
化合物1(0.33g、1.14mmol)をアセトニトリル(10.0mL)に懸濁し、無水酢酸(0.23mL、2.43mmol)、トリエチルアミン(0.67g、4.81mmol)、4−ジメチルアミノピリジン少量を加え、室温で終夜撹拌した。
析出した結晶をろ取、乾燥し、化合物2(0.40g、1.07mmol、93.9%)を得た。
【0132】
1H−NMR(DMSO−d6)δ:7.94(1H,s,H−8),6.76(2H,bs,NH2),6.27(1H,t,H−1’,J=7.00),5.84(2H,bs,NH2),5.60(1H,dd,H−3’,J=4.00,6.80),4.46(1H,d,H−5’a,J=11.5),4.21(1H,d,H−5’b,J=11.5),3.74(1H,s,ethynyl)3.12(1H,m,H−2’a),2.52(1H,m,H−2’b),2.12,2.03(each3H,s,acetyl)
【0133】
(2)3’,5’−ジ−O−アセチル−2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン(化合物3)の合成
【0134】
【化20】
【0135】
化合物2(450mg、1.20mmol)を70%フッ化水素−ピリジン(5.00mL)に溶解し、亜硝酸t−ブチル(0.194mL、1.63mmol)を加え、−10℃で1時間撹拌した。蒸留水を加えた後、クロロホルムで抽出し、有機層を無水硫酸マグネシウム上で乾燥後、減圧下濃縮した。残さにクロロホルム:メタノール(50:1)を加え、析出した結晶をろ取、乾燥し、化合物3(240mg、0.64mmol、53.3%)を得た。
【0136】
1H−NMR(DMSO−d6)δ:8.34(1H,s,H−8),7.94,7.99(each1H,bs,NH2),6.35(1H,t,H−1’,J=6.80),5.68(1H,dd,H−3’,J=5.10,7.05),4.41(1H,d,H−5’a,J=11.6),4.21(1H,d,H−5’b,J=11.6),3.42(1H,s,ethynyl),3.14(1H,m,H−2’a),2.63(1H,m,H−2’b),2.12,2.00(each3H,s,acetyl).
【0137】
(3)2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン(化合物4)の合成
【0138】
【化21】
【0139】
化合物3(200mg、0.53mmol)をメタノール(7.00mL)に溶解し、28%アンモニア水(5.00mL)を加え、室温で4時間撹拌した。反応液を減圧下濃縮後、残さにクロロホルム:メタノール(20:1)を加え、析出した結晶をろ取した。得られた結晶を水から再結晶化し、化合物4(113mg、0.39mmol、73.6%)を得た。
【0140】
1H−NMR(DMSO−d6)δ:8.30(1H,s,H−8),7.87,7.84(each1H,bs,NH2),6.24(1H,dd,H−1’,J=5.05,7.15),5.57(1H,d,3’−OH,J=5.50),5.30(1H,t,5’−OH,J=6.40),4.57(1H,m,H−3’),3.65(1H,m,H−5’a),3.55(1H,m,H−5’b),3.51(1H,s,ethynyl),2.70(1H,m,H−2’a),2.44(1H,m,H−2’b).
【0141】
合成例2:4’−C−シアノ−2’−デオキシ−2−フルオロアデノシン(化合物8)の合成
【0142】
(1)9−(3,5−ジ−O−アセチル−4−C−シアノ−2−デオキシ−β−D−リボ−ペントフラノシル)−2,6−ジアミノプリン(化合物6)の合成
【0143】
【化22】
【0144】
化合物5(122mg、0.418mmol)をアセトニトリル(5.00mL)に懸濁し、無水酢酸(118μl、1.25mmol)、トリエチルアミン(352μl、2.51mmol)、4−ジメチルアミノピリジン少量を加え、室温で終夜撹拌した。析出した結晶をろ取、乾燥し、化合物6(128mg、0.341mmol、81.6%)を得た。
【0145】
1H−NMR(CDCl3)δ:7.54(1H,s,H−8),6.31(1H,t,H−1’,J=7.00),6.06(1H,dd,H−3’,J=5.00,6.50),5.31(2H,bs,NH2),4.95(1H,d,H−5’a,J=11.5),4.80(2H,bs,NH2),4.37(1H,d,H−5’b,J=12.0),3.43(1H,m,H−2’a),2.63(1H,m,H−2’b),2.23,2.12(each3H,s,acetyl).
【0146】
(2)3’,5’−ジ−O−アセチル−4’−C−シアノ−2’−デオキシ−2−フルオロアデノシン(化合物7)の合成
【0147】
【化23】
【0148】
化合物6(118mg、0.314mmol)を70%フッ化水素−ピリジン(2.30mL)に溶解し、亜硝酸t−ブチル(50.0μl、0.427mmol)を加え、−10℃で3時間撹拌した。亜硝酸t−ブチル(10.0μl、85μmol)を追加し、同温度で更に1時間撹拌した。飽和重曹水を加えた後、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄した。有機層を硫酸マグネシウム上で乾燥後、減圧濃縮した。残さをエタノールに加熱溶解後、冷却し、析出した結晶をろ取、乾燥し、化合物7(53.7mg、0.14mmol、45.2%)を得た。
【0149】
1H−NMR(DMSO−d6)δ:8.35(1H,s,H−8),8.00,7.92(each1H,bs,NH2),6.54(1H,t,H−1’,J=7.00),5.83(1H,dd,H−3’,J=4.00,6.50),4.63(1H,d,H−5’a,J=11.5),4.44(1H,d,H−5’b,J=12.0),3.26(1H,m,H−2’a),2.73(1H,m,H−2’b),2.18,2.05(each3H,s,acetyl).
【0150】
(3)4’−C−シアノ−2’−デオキシ−2−フルオロアデノシン(化合物8)の合成
【0151】
【化24】
【0152】
化合物7(53.7mg、0.142mmol)をメタノール(1.90mL)に溶解し、28%アンモニア水(1.30mL)を加え、室温で30分間撹拌した。反応液を減圧下濃縮後、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 10mL、ヘキサン:酢酸エチル=5:1〜酢酸エチル〜酢酸エチル:メタノール=10:1)で精製し、化合物8(30.2mg、0.10mmol、72.3%)を得た。
【0153】
1H−NMR(DMSO−d6)δ:8.31(1H,s,H−8),7.93,7.82(each1H,bs,NH2),6.43(1H,t,H−1’,J=7.00),6.36(1H,bs,3’−OH),5.74(1H,bs,5’−OH),4.70(1H,t,H−3’,J=5.50),3.80(1H,d,H−5’a,J=12.0),3.65(1H,d,H−5’b,J=12.0),2.93(1H,m,H−2’a),2.47(1H,m,H−2’b).
【0154】
合成例3:2−クロロ−2’−デオキシ−4’−C−エチニルアデノシン(化合物9)の合成
【0155】
【化25】
【0156】
亜硝酸ベンジルトリエチルアンモニウム(1.04g、4.36mmol)をジクロロメタン(24.0mL)に溶解し、塩化アセチル(0.40mL、5.63mmol)を加え、0℃で30分間撹拌した。この溶液に、化合物2(340mg、0.91mmol)のジクロロメタン(6.00mL)溶液を加え、0℃で3時間撹拌した。反応液をクロロホルムで希釈した後、有機層を水洗し、無水硫酸マグネシウム上で乾燥、減圧下濃縮した。残さに28%アンモニア水(10.0mL)、メタノール(15.0mL)を加え、室温で終夜撹拌した後、反応液を減圧下濃縮し、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 50mL、クロロホルム:メタノール=20:1〜10:1)で精製した。得られた残さを更にODSカラムクロマトグラフィー(ODS 50mL、5〜10〜15〜20%アセトニトリル)により精製し、化合物9(39.2mg、0.13mmol、14.3%)を得た。
【0157】
1H−NMR(DMSO−d6)δ:8.34(1H,s,H−8),7.84(2H,bs,NH2),6.27(1H,dd,H−1’,J=5.00,7.00),5.60(1H,d,3’−OH,J=5.00),5.33(1H,t,5’−OH,J=6.00),4.56(1H,m,H−3’),3.64(1H,m,H−5’a),3.56(1H,m,H−5’b),3.52(1H,s,ethynyl),2.68(1H,m,H−2’a),2.45(1H,m,H−2’b).
【0158】
合成例4:2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン5’−H−ホスホネート(化合物10)の合成
【0159】
【化26】
【0160】
化合物4(50.0mg、0.171mmol)をピリジン(2.00mL)に溶解し、ホスホン酸(21.0mg,0.25mmol)、ジシクロヘキシルカルボジイミド(106mg、0.51mmol)を加え、室温で5時間撹拌した。沈殿をろ去後、ろ液を減圧下濃縮し、残さを水−クロロホルムで分配した。水層を減圧下濃縮し、残さを分取薄層クロマトグラフィーにより精製した(イソプロパノール:28%アンモニア水:水=7:1:2)。得られた残さをアセトニトリルで共沸後、メタノール−エーテルで粉末とし、化合物10(6.3mg、17.6μmol、10.3%)を得た。
【0161】
1H−NMR(D2O)δ:8.13(1H,s,H−8),6.49(1H,d,H−P,J=645),6.25(1H,dd,H−1’,J=5.00,7.50),3.96(2H,m,H−5’),2.75,2.59(each 1H,m,H−2’).
【0162】
31P−NMR(D2O)δ:6.45.
【0163】
合成例5:2’,3’−ジデヒドロ−2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン(化合物13)の合成
【0164】
【化27】
【0165】
化合物4(0.28g、0.95mmol)をジメチルホルムアミド(7.00mL)に溶解し、t−ブチルクロロジフェニルシラン(0.50mL,1.92mmol)、イミダゾール(0.26g、3.82mmol)を加え、室温で終夜撹拌した。反応液にメタノールを加えた後、減圧下濃縮し、残さを酢酸エチル−水で分配した。有機層を無水硫酸マグネシウム上で乾燥後、減圧下濃縮し、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 100mL、クロロホルム:メタノール=20:1)で精製した。粗製の化合物11(0.38g)を得た。
粗製の化合物11(0.38g)をジクロロメタン(10.0mL)に溶解し、−10℃で無水トリフルオロメタンスルホン酸(0.14mL,0.83mmol)、ピリジン(0.14g、1.71mmol)を加え、同温度で2時間撹拌した。反応液に飽和重曹水を加えた後、クロロホルムで抽出した。有機層を無水硫酸マグネシウム上で乾燥後、減圧下濃縮し、得られた粗トリフラートを精製することなく、次の反応に用いた。
【0166】
粗トリフラートを乾燥テトラヒドロフラン(20.0mL)に溶解し、アルゴン雰囲気下、−78℃で、1M ナトリウムヘキサメチルジシラジド−テトラヒドロフラン溶液(2.50mL、2.50mmol)を加え、同温度で2時間撹拌後、反応液を室温に戻し、終夜撹拌した。反応液に水を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウム上で乾燥後、減圧下濃縮し、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 50mL、クロロホルム:メタノール=50:1〜20:1)で精製し、粗製の化合物12(0.20g)を得た。
得られた粗製の化合物12をテトラヒドロフラン(10.0mL)に溶解し、1M フッ化テトラブチルアンモニウム−テトラヒドロフラン溶液(0.59mL、0.59mmol)を加え、室温で30分間撹拌した。反応液を減圧下濃縮後、クロロホルム:メタノール(10:1)を加え、析出した結晶をろ取した。化合物13(52.0mg、0.19mmol、20.0% from 化合物4)を得た。
【0167】
1H−NMR(DMSO−d6)δ:8.08(1H,s,H−8),7.84(2H,bs,NH2),6.90(1H,t,H−1’,J=1.50),6.43(1H,dd,H−3’,J=2.00,6.00),6.27(1H,dd,H−3’,J=1.00,6.00),5.37(1H,t,5’−OH,J=6.00)3.71(1H,s,ethynyl),3.67(1H,dd,H−5’a,J=6.00,12.0),3.57(1H,dd,H−5’b,J=6.00,12.0).
【0168】
合成例6:2’,3’−ジデヒドロ−2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン5’−H−ホスホネート(化合物14)の合成
【0169】
【化28】
【0170】
化合物13(13.0mg、0.047mmol)をピリジン(0.7mL)に溶解し、ホスホン酸(7.7mg,0.094mmol)、ジシクロヘキシルカルボジイミド(29.2mg、0.14mmol)を加え、室温で1時間撹拌した。反応液を減圧下濃縮し、残さをODSカラムクロマトグラフィー(ODS 10mL、0〜1%アセトニトリル)により精製した。得られた残さをDowex 50Wx8カラム(Na型)に通過させた。通過液を濃縮してえられた残さをメタノール−エーテルで粉末とし、化合物14(4.3mg、12μmol、25.5%)を得た。
【0171】
1H−NMR(MeOD)δ:8.30(1H,s,H−8),6.69(1H,d,H−P,J=625),7.02(1H,bt,H−1’),6.48(1H,dd,H−2’,J=2.00,5.50),6.22(1H,dd,H−3’,J=1.00,5.50),4.18(1H,dd,H−5’a,J=7.50,11.0),3.99(1H,dd,H−5’b,J=7.50,11.0).
【0172】
31P−NMR(MeOD)δ:4.11.
【0173】
合成例7:2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン(化合物23)の合成
【0174】
(1)1,2−O−イソプロピリデン−4−C−トリエチルシリルエチニル−α−D−キシロ−ペンタフラノース(化合物16)の合成
【0175】
【化29】
【0176】
塩化オキサリル(0.54mL、6.19mmol)をジクロロメタン(10.0mL)に溶解後、−60℃でジメチルスルホキシド(0.90mL、12.7mmol)を滴下し、同温度で15分間撹拌した。ここに化合物15(1.06g、3.13mmol、Biosci.Biotech.Biochem.,57,1433−1438(1993))のジクロロメタン溶液(15.0mL)を滴下し、同温度で30分間撹拌した。トリエチルアミン(1.86mL、13.3mmol)を加えた後、反応液を室温に戻し、30分間撹拌した。反応液をクロロホルムで希釈後、水洗し、有機層を無水硫酸マグネシウム上で乾燥した。有機層を減圧下濃縮し、得られた粗アルデヒドを精製することなく次の反応に用いた。
粗アルデヒドをジクロロメタン(40.0mL)に溶解し、0℃で四臭化炭素(2.08g、6.27mmol)、トリフェニルホスフィン(3.28g、12.5mmol)を加えた後、室温で1時間撹拌した。反応液にトリエチルアミン(2.60mL、18.7mmol)を加えた後、クロロホルムで希釈し、有機層を水洗した。有機層を無水硫酸マグネシウム上で乾燥後、減圧下濃縮し、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 100mL、ヘキサン:酢酸エチル=3:1)で精製した。粗ジブロモエテン(1.42g)を得た。
【0177】
粗ジブロモエテン(1.42g、2.89mmol)を乾燥テトラヒドロフラン(20.0mL)に溶解し、アルゴン雰囲気下、−10℃で2.2M メチルリチウム−エーテル溶液(4.49mL、9.88mmol)を加え、同温度で5分間撹拌した。ここにクロロトリエチルシラン(0.95mL、5.66mmol)を加え、更に30分間撹拌した。反応液に飽和塩化アンモニウム水溶液を加え撹拌後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウム上で乾燥後、減圧下濃縮し、得られた粗アルキンを得た。
粗アルキンを酢酸(80.0mL)に溶解し、水(20.0mL)を加え、室温で終夜撹拌した。反応液を減圧下濃縮し、得られた残さをトルエンで共沸した。残さをシリカゲルカラムクロマトグラフィー(シリカゲル 50mL、ヘキサン:酢酸エチル=3:1)で精製し、化合物16(0.70g、2.13mmol、64.4%)を得た。
【0178】
1H−NMR(CDCl3)δ:6.00(1H,d,H−1,J=3.50),4.60(1H,d,H−2,J=4.00),4.58(1H,d,H−3,J=5.00),3.96−3.91(3H,m,H−5 and 3−OH),2.50(1H,t,5−OH),1.64,1.33(each3H,s,acetonide),0.97(9H,t,Et,J=8.00),0.59(6H,q,Et,J=8.00).
【0179】
(2)5−O−t−ブチルジフェニルシリル−1,2−O−イソプロピリデン−4−C−トリエチルシリルエチニル−α−D−キシロ−ペンタフラノース(化合物17)の合成
【0180】
【化30】
【0181】
化合物16(0.70g、2.13mmol)をジメチルホルムアミド(3.50mL)に溶解し、t−ブチルクロロジフェニルシラン(0.66mL、2.54mmol)、イミダゾール(0.35g、5.14mmol)を加え、終夜撹拌した。反応液にメタノールを加え減圧下濃縮し後、残さを酢酸エチルに溶解した。有機層を水洗後、無水硫酸マグネシウム上で乾燥し、減圧下濃縮した。残さをシリカゲルカラムクロマトグラフィー(シリカゲル 100mL、ヘキサン:酢酸エチル=5:1)で精製し、化合物17(1.10g、1.94mmol、91.1%)を得た。
【0182】
1H−NMR(CDCl3)δ:7.72−7.36(10H,s,aromatic),6.02(1H,d,H−1,J=3.50),4.66(1H,d,H−3,J=5.50),4.63(1H,d,H−1,J=4.00),4.05(1H,d,H−5a,J=10.5),3.99(1H,d,3−OH,J=5.50),3.93(1H,d,H−5’b,J=10.5),1.65,1.35(each3H,s,acetonide),1.06(9H,s,t−Bu),0.93(9H,t,Et,J=8.00),0.56(6H,q,Et,J=8.00).
【0183】
(3)5−O−t−ブチルジフェニルシリル−3−デオキシ−1,2−O−イソプロピリデン−4−C−トリエチルシリルエチニル−α−D−キシロ−ペントフラノース(化合物18)の合成
【0184】
【化31】
【0185】
化合物17(1.10g、1.94mmol)をアセトニトリル(20.0mL)に溶解し、クロロチオノギ酸フェニル(0.40mL、2.89mmol)、4−ジメチルアミノピリジン(0.71g、5.81mmol)を加え、室温で3時間撹拌した。反応液を酢酸エチルで希釈後、有機層を0.1N塩酸、飽和重曹水で洗浄し、無水硫酸マグネシウム上で乾燥した。有機層を減圧下濃縮し、得られた粗チオ炭酸エステルを精製することなく次の反応に用いた。
粗チオ炭酸エステルをトルエンで3回共沸した後、トルエン(30.0mL)に溶解し、減圧脱気した。この溶液をアルゴン雰囲気下、80℃で水素化トリブチルスズ(2.61mL、9.70mmol)、少量のアゾビス(イソブチロニトリル)を加え、同条件で1時間撹拌した。反応液を減圧下濃縮後、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 100mL、ヘキサン:酢酸エチル=10:1)で精製し、化合物18(1.07g、1.94mmol、quant.)を得た。
【0186】
1H−NMR(CDCl3)δ:7.69−7.38(10H,m,aromatic),5.90(1H,d,H−1,J=4.00),4.85(1H,t,H−2,J=5.00),3.82(1H,d,H−5a,J=11.0),3.58(1H,d,H−5b,J=10.5),2.64(1H,dd,H−3a,J=6.00,14.0),2.40(1H,d,H−3b,J=14.0),1.68,1.36(each3H,s,acetonide),1.04(9H,s,t−Bu)0.92(9H,t,Et,J=8.00),0.54(6H,q,Et,J=8.00).
【0187】
(4)1,2−ジ−O−アセチル−5−O−t−ブチルジフェニルシリル−3−デオキシ−4−C−トリエチルシリルエチニル−D−キシロ−ペントフラノース(化合物19)の合成
【0188】
【化32】
【0189】
化合物18(1.07g、1.94mmol)を80%酢酸(100mL)に溶解し、トリフルオロ酢酸(10.0mL)を加え、40℃で3時間撹拌した。反応液を減圧下濃縮後、残さをトルエンで共沸し、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 100mL、ヘキサン−酢酸エチル=4:1)で精製した。得られた残さをピリジン(20.0mL)に溶解し、無水酢酸(0.49mL)を加え、室温で終夜撹拌した。反応液を減圧下濃縮後、残さをトルエンで共沸し、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 100mL、ヘキサン:酢酸エチル=5:1)で精製した。化合物19(0.75g、)を得た。
【0190】
1H−NMR(CDCl3)δ:7.71−7.37(10H,m,aromatic),6.44(0.3H,d,H−1−alpha,J=4.50),6.30(0.7H,s,H−1−beta),5.36(0.3H,m,H−2−alpha),5.19(0.7H,d,H−2−beta,J=5.50),3.77,3.74(each0.7H,d,H−5−beta,J=10.0),3.76,3.62(each0.3H,d,H−5−alpha,J=11.0),2.86(0.3H,dd,H−3a−alpha,J=8.50,12.5),2.72(0.7H,dd,H−3a−beta,J=5.50,14.0),2.39(0.3H,dd,H−3b−alpha,J=10.0,12.5),2.33(0.7H,d,H−3b−beta,J=14.0),2.11,2.08(each0.9H,s,acety−alpha),2.10,1.80(each2.1H,s,acetyl−beta),1.074(6.3H,s,t−Bu−beta),1.067(2.7H,s,t−Bu−alpha),0.97(6.3H,t,Et−beta,J=8.00),0.94(2.7H,t,Et−alpha,J=8.00),0.58(4.2H,q,Et−beta,J=8.00),0.54(1.8H,q,Et−alpha,J=8.00).
【0191】
(5)9−(2−O−アセチル−5−O−t−ブチルジフェニルシリル−3−デオキシ−4−C−トリエチルシリルエチニル−β−D−キシロ−ペントフラノシル)−2,6−ジアミノプリン(化合物20)の合成
【0192】
【化33】
【0193】
2,6−ジアミノプリン(1.21g、8.06mmol)をアセトニトリル(24.0mL)に懸濁し、N,O−ビス(トリメチルシリル)アセトアミド(11.9mL、48.1mmol)を加え、80℃で3時間撹拌した。この溶液を減圧下濃縮後、残さを1,2−ジクロロエタンで3回共沸した。残さに化合物19(2.39g、4.02mmol)の1,2−ジクロロエタン溶液(24.0mL)、トリフルオロメタンスルホン酸トリメチルシリル(3.05mL、16.9mmol)を加え、アルゴン雰囲気下、50℃で5時間、80℃で10時間撹拌した。飽和重曹水を加え撹拌後、溶液をセライトろ過した。有機層を無水硫酸マグネシウム上で乾燥後、減圧下濃縮し、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 300mL、クロロホルム:メタノール=20:1)で精製した。ヘキサン−酢酸エチルから結晶化し、化合物20(1.60g、2.34mmol、58.2%)を得た。
【0194】
1H−NMR(CDCl3)δ:7.67−7.33(10H,m,aromatic),6.13(1H,d,H−1’,J=3.50),5.77(1H,md,H−2’),5.28(2H,bs,NH2),4.49(2H,bs,NH2),3.67(1H,d,H−5’a,J=10.5),3.79(1H,d,H−5’b,J=11.0),3.18(1H,dd,H−3’a,J=7.50,14.0),2.37(1H,dd,H−3’b,J=3.00,14.0),1.06(9H,s,t−Bu),0.99(9H,t,Et,J=8.00),0.61(6H,q,Et,J=8.00).
【0195】
(6)5’−O−t−ブチルジメチルシリル−3’−デオキシ−4’−C−トリエチルシリルエチニル−2−フルオロアデノシン(化合物21)の合成
【0196】
【化34】
【0197】
化合物20(100mg、0.146mmol)をピリジン(3.00mL)に溶解し、−15℃でフッ化水素−ピリジン(7.00mL)、亜硝酸t−ブチル(160μl、1.34mmol)を加え、同温度で30分間撹拌した。反応液に水を加えた後、酢酸エチルで抽出した。有機層を飽和重曹水で洗浄後、無水硫酸マグネシウム上で乾燥し、減圧下濃縮した。残さをシリカゲルカラムクロマトグラフィー(シリカゲル 300mL、クロロホルム:メタノール=100:1〜20:1)で精製した。得られた残さ(48.9mg)をジクロロメタン(1.30mL)に溶解し、0℃でトリフルオロメタンスルホン酸t−ブチルジメチルシリル(36.0μl、0.157mmol)、コリジン(44.1μl、0.0331mmol)を加え、同温度で50分間撹拌した。反応液に水を加え、クロロホルムで抽出した。有機層を0.01N塩酸、飽和重曹水で洗浄後、無水硫酸マグネシウム上で乾燥し、減圧下濃縮した。残さをジオキサン(3.00mL)に溶解し、28%アンモニア水(0.30mL)を加え、室温で30分間撹拌した。反応液を減圧下濃縮後、残さをメタノール(1.20mL)に溶解し、28%アンモニア水(0.80mL)を加え、室温で2時間撹拌した。反応液を減圧下濃縮し、析出した結晶をろ取し、化合物21(34.0mg、0.0652mmol、44.7%)を得た。
【0198】
1H−NMR(CDCl3)δ:8.04(1H,s,H−8),5.99(1H,d,H−1’,J=4.00),5.80(2H,bs,NH2),4.73(1H,m,H−2’),4.23(1H,d,2’−OH,J=5.00),3.88(1H,d,H−5’a,J=11.0),3.70(1H,d,H−5’b,J=11.0),2.82(1H,dd,H−3’a,J=7.50,13.0),2.42(1H,dd,H−3’b,J=6.50,13.0),1.01(9H,t,Et,J=8.00),0.80(9H,s,t−Bu),0.64(6H,q,Et,J=8.00),0.039,−0.013(each3H,s,Me).
【0199】
(7)5’−O−t−ブチルジメチルシリル−2’,3’−ジデオキシ−4’−C−トリエチルシリルエチニル−2−フルオロアデノシン(化合物22)の合成
【0200】
【化35】
【0201】
化合物21(32.0mg、0.061mmol)をアセトニトリルで3回共沸後、アセトニトリル(1.00mL)に溶解し、クロロチオノギ酸フェニル(12.7μl、0.092mmol)、4−ジメチルアミノピリジン(22.5mg、0.180mmol)を加え、室温で1時間撹拌した。反応液を酢酸エチルで希釈後、有機層を0.01N塩酸、飽和重曹水で洗浄し、無水硫酸マグネシウム上で乾燥した。有機層を減圧下濃縮し、得られた粗チオ炭酸エステルを精製することなく次の反応に用いた。
粗チオ炭酸エステルをトルエンで3回共沸した後、トルエン(1.00mL)に溶解し、減圧脱気した。この溶液をアルゴン雰囲気下、80℃でトリス(トリメチルシリル)シラン(94.6μl、0.306mmol)、少量のアゾビス(イソブチロニトリル)を加え、同条件で1時間撹拌した。反応液を減圧下濃縮後、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 10mL、クロロホルム:メタノール=200:1〜100:1)で精製し、化合物22(26.1mg、0.0516mmol、84.6%)を得た。
【0202】
1H−NMR(CDCl3)δ:8.24(1H,s,H−8),6.36(1H,dd,H−1’,J=2.50,7.00),5.91(2H,bs,NH2),4.04(1H,d,H−5’a,J=11.0),3.81(1H,d,H−5’b,J=11.0
),2.83(1H,m,H−2’a),2.54(1H,m,H−3’a),2.37(1H,m,H−2’b),2.11(1H,m,H−3’b),1.00(9H,t,Et,J=8.00),0.93(9H,s,t−Bu),0.62(6H,q,Et,J=8.00),0.13(6H,s,Me).
【0203】
(8)2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン(化合物23)の合成
【0204】
【化36】
【0205】
化合物22(101mg、0.200mmol)をテトラヒドロフラン(10mL)に溶解し、1M フッ化テトラブチルアンモニウム−テトラヒドロフラン溶液(0.42mL、0.42mmol)を加え、室温で5分間撹拌した。反応液に酢酸(24μl)を加えた後、減圧下濃縮し、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 15mL、クロロホルム:メタノール=40:1〜20:1)で精製した。化合物23(53.0mg、0.191mmol、95.7%)を得た。
【0206】
1H−NMR(MeOD)δ:8.23(1H,s,H−8),6.22(1H,dd,H−1’,J=4.00,7.00),3.77(1H,d,H−5’a,J=12.5),3.61(1H,d,H−5’b,J=12.0),2.94(1H,s,ethynyl),2.66(1H,m,H−2’a),2.54(1H,m,H−3’a),2.42(1H,m,H−2’b),2.11(1H,m,H−3’b).
【0207】
合成例8:2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン5’−H−ホスホネート(化合物24)の合成
【0208】
【化37】
【0209】
化合物23(20.0mg、0.07mmol)をピリジン(1mL)に溶解し、ホスホン酸(11.8mg,0.144mmol)、ジシクロヘキシルカルボジイミド(44.7mg、0.216mmol)を加え、室温で5時間撹拌した。反応液を減圧下濃縮し、残さをODSカラムクロマトグラフィー(ODS 10mL、0−1%アセトニトリル)により精製した。得られた残さをDowex 50Wx8カラム(Na型)に通過させた。通過液を濃縮してえられた残さをメタノール−エーテルで粉末とし、化合物24(4.7mg、13μmol、18.6%)を得た。
【0210】
1H−NMR(MeOD)δ:8.37(1H,s,H−8),6.77(1H,d,H−P,J=625),6.32(1H,dd,H−1’,J=4.00,6.50),4.09(1H,m,H−5’),2.71(2H,m,H−2’a,H−3’a,),2.52(1H,m,H−2’b),2.29(1H,m,H−3’b).
【0211】
31P−NMR(MeOD)δ:4.52
【0212】
製剤例1:錠剤
本発明化合物 30.0mg
微粉末セルロース 25.0mg
乳糖 39.5mg
スターチ 40.0mg
タルク 5.0mg
ステアリン酸マグネシウム 0.5mg
上記組成から常法によって錠剤を調製する。
【0213】
製剤例2:カプセル剤
本発明化合物 30.0mg
乳糖 40.0mg
スターチ 15.0mg
タルク 5.0mg
上記組成から常法によってカプセル剤を調製する。
【0214】
製剤例3:注射剤
本発明化合物 30.0mg
グルコース 100.0mg
上記組成を注射用精製水に溶解して注射剤を調製する。
【0215】
以下に試験例を示す。試験においては薬剤として以下の本発明化合物と公知化合物を使用した。
本発明化合物:
化合物4: 2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン
化合物8: 4’−C−シアノ−2’−デオキシ−2−フルオロアデノシン
化合物9: 2−クロロ−2’−デオキシ−4’−C−エチニル−アデノシン
化合物10:2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン5’−H−ホスホネート
化合物13:2’,3’−ジデヒドロ−2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン
公知化合物:
AZT:アジドチミジン
EdAdo:2’−デオキシ−4’−C−エチニルアデノシン
EdDAP:9−(4−C−エチニル−2−デオキシ−リボペントフラノシル)−2,6−ジアミノプリン
ddAdo:2’,3’−ジデオキシアデノシン
【0216】
試験例1
<方法>抗ヒト免疫不全ウイルス(HIV)活性
1)MT−4細胞を用いたMTT法
1.96ウエルプレートを用いて被験薬剤を希釈する(100μl)。ここに1ウエルあたり10000個になるようにHIV−1(IIIbstrain;100TCID50)感染及び非感染MT−4細胞を加えて、37℃で5日間培養する。
2.MTT(20μl、7.5mg/mL)を加えて更に2〜3時間培養する。
3.培養終了後、120μlをとり、MTT停止液(Triton X−100 4%、HCl 0.04Nを加えたイソプロパノール(Isopropanol))を加え、撹拌して生成したホルマザン(formazam)を溶解し、540nmにおける吸光度を測定する。この測定値は生細胞数に比例するため、感染MT−4細胞を用いた試験の測定値が50%になった被験薬剤の濃度をEC50、また非感染MT−4細胞を用いた試験の測定値が50%になった被験薬剤の濃度をCC50とする。
【0217】
2)HeLa CD4/LTR−beta−Gal細胞を用いたMAGIアッセイ
1.HeLa CD4/LTR−beta−Gal細胞を1ウエルあたり10000個の割合で96ウエルに加える。12〜24時間後に培養液を捨て、希釈した被験薬剤(100μl)を加える。
2.各種HIV株(野生株:WT、耐性株:MDR、M184V;各50TCID50相当)を加えて48時間培養する。
3.培養終了後、1%ホルムアルデヒド(formaldehyde)、0.2%グルタールアルデヒド(glutaraldehyde)を加えたPBSで細胞を5分間固定する。
4.PBSで3回洗浄後、0.4mg/mL X−Galで1時間染色し、青く染色された細胞の数を透過型実体顕微鏡下で各ウエルのプラーク数を数え、青染された細胞を50%減少させる被験薬剤の濃度をEC50とする。
【0218】
<結果>抗ヒト免疫不全ウイルス(HIV)活性及び細胞毒性
1)MT−4細胞を用いたMTT法
【0219】
【表1】
【0220】
2)HeLa CD4/LTR−beta−Gal細胞を用いたMAGIアッセイ
【0221】
【表2】
【0222】
試験例2
<方法>化合物4のアデノシンデアミナーゼに対する安定性
0.5mMの化合物4(50mM Tris−HCl緩衝液溶液(pH7.5))0.5mLに子ウシ腸管由来アデノシンデアミナーゼ 0.01unitを加え、25℃でインキュベートする。
15分ごとに反応液5μlを取り、高速液体クロマトグラフィー(HPLC)により分析する。反応時間0分のときの被験薬剤のピーク面積を100%として、その経時変化を観察する。なお、HPLC分析は、以下の条件で行った。
カラム:YMC−Pack ODS−A(250×6.0mm)
溶出液:15% MeCN−50mM TEAA
流速: 1mL/min
温度: 30℃
検出: 260nm
【0223】
<結果>
その結果、図1に示すように、従来の2’−デオキシ−4’−C−エチニルアデノシン(EdAdo)は脱アミノされるに対し、本発明化合物の2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン(化合物4)は全く脱アミノされず、アデノシンデアミナーゼに対し、抵抗性を有することが明らかとなった。
【0224】
試験例3
<方法>化合物4の酸性条件下における安定性
化合物4(2.9mg)もしくは2’,3’−ジデオキシアデノシン(ddAdo:2.4mg)を予め37℃に暖めた試験液(塩化ナトリウム2.0gに塩酸7.0mL及び水を加えて溶かし1000mLとしたもの)10mLに溶解し、同温度でインキュベートする。
反応液100μlを取り、0.1規定水酸化ナトリウム水溶液で中和後、5μlをHPLCにより分析する。なお、HPLC分析条件は、試験例2と同じである。
【0225】
<結果>
その結果、従来のddAdoは本条件下5分ほどで約98%が分解されるのに対し(図3)、本発明化合物の2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン(化合物4)の分解は非常に遅く、酸性条件下比較的安定であることが明らかとなった(図2)。
【0226】
試験例4
<方法>化合物4のin vivoにおける急性毒性試験
6週齢、雄性、ICRマウスに被検薬剤(化合物4:生理食塩水に溶解又は懸濁)を一群8匹ずつ、100mg/kgまで経口あるいは静脈内投与し、7日間マウスの生死、体重を測定した。
【0227】
<結果>
化合物4は、単回投与では経口、静脈内いずれの投与経路でも100mg/kgまでマウスの死亡は認められなかった(下記表3)。また、図4に示すように体重減少は見られず、下痢のような症状も観察されなかったことから、本発明化合物の2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン(化合物4)はマウスにおいて急性毒性を示さないことが明らかになった。
【0228】
【表3】
【技術分野】
【0001】
本発明は、4’−C−置換−2−ハロアデノシン誘導体及びその医薬用途、特にエイズ(AIDS)の治療用用途に関するものである。
【背景技術】
【0002】
AZT(ジドブジン)、ddI(ジダノシン)、ddC(ザルシタビン)、d4T(スタブジン)、3TC(ラミブジン)などのヌクレオシド系逆転写酵素阻害剤(nucleoside reverse transcriptase inhibitors;NRTIs)にプロテアーゼ阻害剤(protease inhibitors;PIs)を加えた、いわゆる「強力な抗レトロウイルス剤による化学療法(highly active antiretroviral therapy;HAART)」と呼ばれる多剤併用療法によって、AIDSの臨床像は一変し、AIDSによる死亡者数も各国で激減した。
【0003】
しかし、HAARTによりAIDSによる死亡者が激減する一方で、複数の薬剤に対して交叉耐性を示す多剤耐性HIV−1(human immunodeficiency virus−1)株が出現し、たとえば、AZTと3TCの両方に耐性のHIVに感染した患者は、1990年初頭ではほとんど見られなかったのに対し、1995〜1996年になると42%にものぼっていることが報告されている。
【0004】
大類らは、2’−デオキシ−4’−C−エチニルヌクレオシドを合成し、それら化合物の抗HIV活性を測定した結果、特定の構造を有する2’−デオキシ−4’−C−エチニルヌクレオシドがAZTと同等あるいはAZTを凌ぐ優れた抗HIV活性を有すること、AZT、ddI、ddC、d4T、3TCなどの複数の抗HIV剤に耐性を有する多剤耐性ウイルス株にも有効なことを明らかにした(非特許文献1〜8、特許文献1〜3)。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】WO00/69876
【特許文献2】WO00/69877
【特許文献3】WO03/68796
【非特許文献】
【0006】
【非特許文献1】Nucleic Acids Symp Ser,Jan 2000;(44):105−6.
【非特許文献2】J Med Chem,Nov 2000;43(23):4516−25
【非特許文献3】Curr Drug Targets Infect Disord,May 2001;1(1):1−10
【非特許文献4】Antimicrob.Agents Chemother.,May 2001;45:1539−1546
【非特許文献5】Nucleosides Nucleotides Nucleic Acids,May 2003;22(5−8):887−9.
【非特許文献6】Chem.Pharm.Bull.,42(1994),p1688
【非特許文献7】J.Med.Chem.,39(1996),p3847
【非特許文献8】Bioorg Med Chem Lett,Nov 2003;13(21):3775−7
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明者らは、in vitroにおいて、種々の4’−C−置換ヌクレオシドの中でも特に優れた抗HIV活性を示した4’−C−エチニルプリンヌクレオシド誘導体及び4’−C−シアノプリンヌクレオシド誘導体について毒性の評価を行った。その結果、(1)最も強い抗HIV活性を示す2,6−ジアミノプリン誘導体及びグアニン誘導体はin vitro、in vivoにおいて毒性を示すこと、(2)毒性の低いアデニン誘導体は血中でアデノシンデアミナーゼにより速やかにヒポキサンチン誘導体へと変換され、抗HIV活性が減弱することが明らかとなった。
【課題を解決するための手段】
【0008】
本発明者らは、選択係数(細胞毒性濃度/抗HIV活性濃度)の更なる向上とアデノシンデアミナーゼによる不活性化に対する耐性付与を目的とし、種々の4’−C−置換プリンヌクレオシドの中で、強い抗HIV活性を示し、かつ毒性の低い4’−C−置換−2’−デオキシアデノシンをリード化合物とし、このリード化合物に化学的修飾を施すことにより数多くの誘導体を合成した。
【0009】
従来、アデノシンデアミナーゼによる不活性化に対する耐性付与に関しては、アデノシン誘導体の塩基部2位に電子吸引性のハロゲン原子を導入することにより、アデノシンデアミナーゼに対して若干の抵抗性を示すようになることが知られている(非特許文献6と7)が、ハロゲン原子の導入による選択係数の向上に関しては何ら示唆されていない。
【0010】
唯一、d4T(スタブジン;2’,3’−ジデヒドロ−3’−デオキシチミジン)の4’位にエチニル基を導入することで選択係数の向上が報告されているものの(非特許文献8)、基本骨格を著しく異にするプリン系ヌクレオシドであるアデノシン誘導体で同様の効果を期待することはできず、何ら参考になるものではなかった。
【0011】
新たに合成した誘導体の抗HIV活性等を検討した結果、リード化合物である2’−デオキシ−4’−C−エチニルアデノシンの塩基部2位にフッ素原子を導入した2’−デオキシ−4’−C−エチニル−2−フルオロアデノシンは、アデノシンデアミナーゼによる不活性化に対する耐性付与だけでなく、AZT、ddI、ddC、d4T、3TCなどの複数の抗HIV剤に耐性を有する多剤耐性ウイルス株にも有効で、しかも抗HIV活性が増強され、一方で、その細胞毒性は著しく減少することが明らかとなった。
【0012】
したがって、本発明は、この知見を更に発展させ、2−ハロアデニンを核酸塩基とし、糖部4位にエチニル基又はシアノ基を有する種々の4’−C−置換−2−ハロアデノシン誘導体を合成し、その活性を確認することにより完成されたものである。
【0013】
すなわち、本発明は、下記式[I]、[II]又は[III]で表される4’−C−置換−2−ハロアデノシン誘導体及び当該誘導体と薬学的に許容される担体とを含有してなる医薬組成物に関するものである。
【0014】
【化1】
【0015】
(式中、Xはハロゲン原子、R1はエチニル基又はシアノ基、R2は、水素原子、リン酸又はその誘導体残基を示す。)
また、本発明は、上記4’−C−置換−2−ハロアデノシン誘導体又はそれを含む医薬組成物をヒトを含む動物に投与することを特徴とするエイズの治療方法に関するものである。
【発明の効果】
【0016】
本発明化合物は、後述試験例に示すように、たとえば、2’−デオキシ−4’−C−エチニル−2−フルオロアデノシンは、アデノシンデアミナーゼによる不活性化に対する耐性付与だけでなく、AZT、ddI、ddC、d4T、3TCなどの複数の抗HIV剤に耐性を有する多剤耐性ウイルス株にも有効で、しかもリード化合物である2’−デオキシ−4’−C−エチニルアデノシンの抗HIV活性と比較して144倍と予想できないほどに抗HIV活性が増強され、一方で、その細胞毒性は著しく減少することが明らかとなった。この結果、この化合物の選択係数は110,000となり、従来の2’−デオキシ−4’−C−エチニルアデノシン(EdAdo)の1,630を遙かに上回る驚くべき結果となった。
このように、本発明化合物は、優れた抗HIV作用、特にAZT、DDI、DDC、D4T、3TCなどの抗HIV剤の複数の薬剤に耐性を有する多剤耐性HIV株にも有効で、細胞毒性も低く、アデノシンデアミナーゼによる不活性化に対する耐性も付与されていることから、医薬品、特にエイズ治療薬として有用である。
【図面の簡単な説明】
【0017】
【図1】アデノシンデアミナーゼによる脱アミノ化反応における化合物の安定性を示したものである。黒四角は本発明化合物2’−デオキシ−4’−C−エチニル−2−フルオロアデノシンの結果を、黒丸は公知の2’−デオキシ−4’−C−エチニルアデノシンの結果を示したものである。
【図2】酸性条件下における本発明化合物2’−デオキシ−4’−C−エチニル−2−フルオロアデノシンの安定性を示したものである。
【図3】酸性条件下における公知の2’,3’−ジデオキシアデノシン(ddAdo)の安定性を示したものである。
【図4】本発明化合物2’−デオキシ−4’−C−エチニル−2−フルオロアデノシンを投与後のマウスの体重変化を示したものである。図4中のグラフAは経口投与時の結果を、グラフBは静脈内投与時の結果を示す。なお、各グラフ中の丸印はプラセボを示し、三角印、四角印は投与量がそれぞれ30mg/kg、100mg/kgのときの結果を示す。
【発明を実施するための形態】
【0018】
(1)化合物
本発明化合物は、前記式[I]、[II]又は[III]で表されるものである。上記式中、R2のリン酸又はその誘導体残基としては、リン酸残基、ジリン酸残基、トリリン酸残基、ホスホン酸残基、及びこれらのジエステル、トリエステル等のポリエステル、モノアミデート、ジアミデート等のアミデート、チオエート、セレノエート、ボラノエート等を例示することができる。また、Xのハロゲン原子としては、臭素原子、ヨウ素原子、フッ素原子又は塩素原子を例示することができる。
【0019】
このような化合物のうち、(a)R2が水素原子又はホスホン酸残基、(b)Xがフッ素原子又は塩素原子、(c)R1がエチニル基である条件の1つ又は複数を満たすものを好適な化合物として例示することができる。具体的には、以下の化合物を挙げることができる。
【0020】
<式[I]化合物>
2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン、4’−C−シアノ−2’−デオキシ−2−フルオロアデノシン、2−クロロ−2’−デオキシ−4’−C−エチニルアデノシン又は2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン5’−H−ホスホネート
【0021】
<式[II]化合物>
2’,3’−ジデヒドロ−2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン、2’,3’−ジデヒドロ−2’,3’−ジデオキシ−4’−C−シアノ−2−フルオロアデノシン、2’,3’−ジデヒドロ−2’,3’−ジデオキシ−4’−C−エチニル−2−クロロアデノシン又は2’,3’−ジデヒドロ−2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン5’−H−ホスホネート
【0022】
<式[III]化合物>
2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン、2’,3’−ジデオキシ−4’−C−シアノ−2−フルオロアデノシン、2’,3’−ジデオキシ−4’−C−エチニル−2−クロロアデノシン又は2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン5’−H−ホスホネート
【0023】
本発明化合物は、塩、水和物又は溶媒和物の形態であってもよい。そのような塩としては、R2が水素原子である場合には塩酸塩又は硫酸塩などの酸付加物、R2がリン酸残基である場合にはナトリウム塩、カリウム塩又はリチウム塩などのアルカリ金属塩、カルシウム塩などのアルカリ土類金属塩もしくはアンモニウム塩などの薬学的に許容される任意の塩が例示される。
また、水和物又は溶媒和物としては、本発明の化合物又はその塩1分子に対し、0.1〜3.0分子の水又は溶媒が付着したものを例示することができる。更に、本発明の化合物には、互変異性体などの各種異性体も包含されうる。
【0024】
(2)製造法
本発明化合物[I]は、以下に説明する工程により製造することができる。
第1工程;
第1工程は式[IV]で表される化合物の3’位水酸基及び5’位水酸基を保護し、式[V]で表される化合物を得る工程である。
【0025】
【化2】
【0026】
(式中、Pは保護基,R1はエチニル基又はシアノ基を示す。)
【0027】
式[IV]で表される原料化合物のうち、R1がエチニル基である化合物はJ.Med.Chem.,43,4516−4525(2000)に記載されており、R1がシアノ基である化合物はWO03/68796に記載されており、公知である。
Pで表される3’,5’位水酸基の保護基としては、水酸基の保護基として常用されているものであればよく、たとえばエーテル系保護基、アシル系保護基、シリル系保護基、アセタール系保護基などを例示することができる。より具体的には、エーテル系保護基としては、メチルエーテル、第3級ブチルエーテル、ベンジルエーテル、メトキシベンジルエーテル、トリチルエーテルなどを、アシル系保護基としてはアセチル、ベンゾイル、ピバロイルなどを、シリル系保護基としてはt−ブチルジメチルシリル、t−ブチルジフェニルシリル、トリメチルシリル、トリエチルシリルなどを、アセタール系保護基としてはイソプロピリデン、エチリデン、メチリデン、ベンジリデン、テトラヒドロピラニル、メトキシメチルなどをそれぞれ使用することができる。
【0028】
保護基の導入は常法によって行うことができる。たとえば、ピリジン、アセトニトリル、ジメチルホルムアミド等の有機溶媒中、アルカリ金属アルコキシド、トリエチルアミン、4−ジメチルアミノピリジン、イミダゾール等の塩基の存在下、式(IV)の化合物と保護基導入試薬(例えば、アルキルハライド、酸ハライド、酸無水物、アルキルシリルハライド等)とを、−10〜100℃で反応させることによって実施することができる。
【0029】
第2工程;
第2工程は、式[V]で表される化合物の2位アミノ基をハロゲン原子へと変換し、式[VI]で表される化合物を得る工程である。
【0030】
【化3】
【0031】
(式中、Pは保護基、Xはハロゲン原子、R1はエチニル基又はシアノ基を示す。)
【0032】
式[VI]で表される化合物は、式[V]で表される化合物の2位アミノ基を亜硝酸誘導体で処理後、ハロゲン化試薬を用いて塩基部2位にハロゲン原子を導入するか、又は、2,6位のアミノ基を同条件下処理し、2,6−ジハロプリン誘導体とした後、アンモニア処理により塩基部6位のハロゲン原子をアミノ基とすることで合成することができる。
【0033】
式[V]で表される化合物の2位アミノ基をフッ素へと置換する試薬としては、テトラフルオロホウ酸中の亜硝酸ナトリウム、フッ化水素−ピリジン中の亜硝酸t−ブチル等の亜硝酸エステルなどを例示することができる。
【0034】
反応条件は用いる試薬により異なり、たとえば、フッ化水素−ピリジン中、亜硝酸t−ブチルを用いる場合、フッ化水素−ピリジンを溶媒として、亜硝酸t−ブチルを1〜3モル用い、−50℃から室温で15分から5時間程度反応させればよい。また、2,6−ジフルオロプリン誘導体とした場合は、引き続き、ジオキサン、メタノール等の有機溶媒中、アンモニア水処理すればよい。
【0035】
一方、式[V]で表される化合物の2位アミノ基を塩素へと置換する試薬としては、ジクロロメタンなどの有機溶媒中、三塩化アンチモン−亜硝酸t−ブチル、塩化アセチル−亜硝酸ベンジルトリエチルアンモニウムの組み合わせなどを例示することができる。
【0036】
反応条件は用いる試薬により異なり、たとえば、塩化アセチル−亜硝酸ベンジルトリエチルアンモニウムの組み合わせを用いる場合、ジクロロメタン等の有機溶媒中、化合物[V]1モルに対して、亜硝酸ベンジルトリエチルアンモニウム1〜5モルと1〜5モルの塩化アセチルとを−50℃から室温で30分間から3時間程度処理した後、化合物[V]を−50℃から室温で1時間から数日間反応させればよい。また、2,6−ジクロロプリン誘導体とした場合は、引き続き、ジオキサン、メタノール等の有機溶媒中、アンモニア水処理すればよい。
【0037】
こうして得られた化合物[VI]の保護基を除去して、R2が水素である本発明化合物を得、必要に応じてリン酸化して本発明化合物を得る。
【0038】
【化4】
【0039】
(式中、Pは保護基、Xはハロゲン原子、R1はエチニル基又はシアノ基、R2は水素原子、リン酸又はその誘導体残基を示す。)
保護基の除去は、使用した保護基に応じ、酸性加水分解、アルカリ性加水分解、フッ化テトラブチルアンモニウム処理、接触還元などの通常の処理方法から適宜選択して行えばよい。
【0040】
【化5】
【0041】
(式中、Xはハロゲン原子、R1はエチニル基又はシアノ基、R2は水素を示す。)
【0042】
本発明化合物の5’−H−ホスホネート誘導体[VII]を得るには、有機溶媒中、適当な縮合剤を用い、R2が水素である化合物[I]とホスホン酸を縮合すればよい。用いる有機溶媒として、ピリジン、又はトリエチルアミン等の塩基存在下でのジメチルホルムアミド、縮合剤としては、ジシクロヘキシルカルボジイミド、ジイソプロピルカルボジイミド、水溶性カルボジイミド等のカルボジイミド、塩化トルエンスルホニル等のスルホン酸ハロゲン化物、塩化ジフェニルリン酸等のリン酸塩化物を例示することができる。
【0043】
反応条件は用いる試薬により異なり、たとえば、ピリジン中、ジシクロヘキシルカルボジイミドを用いる場合、化合物[I]1モルに対して、ホスホン酸を1〜5モル、ジシクロヘキシルカルボジイミドを1〜10モル用い、0℃〜50℃で1〜24時間程度反応させることにより実施できる。
【0044】
また、R2がモノリン酸残基である化合物を得る場合には、R2が水素原子である化合物をオキシ塩化リン、テトラクロロピロリン酸などのヌクレオシドの5’位の選択的なリン酸化に使用されるリン酸化剤と反応させることにより、更に、R2がジリン酸、トリリン酸残基である化合物を得るには、対応する5’−モノリン酸化合物をホスホイミダゾリド、ホスホモルホリデート、ジフェニルリン酸無水物などを用いて活性化した後、それぞれ、リン酸、ピロリン酸、又はこれらの適当な塩と反応させることにより、遊離酸型又は塩型の目的化合物を得ることができる。
【0045】
本発明化合物[II]は、以下に説明する工程により製造することができる。
第1工程;
第1工程は式[I]で表され、R2が水素である化合物の5’位水酸基を選択的に保護し、式[VIII]で表される化合物を得る工程である。
【0046】
【化6】
【0047】
(式中、Pは保護基、Xはハロゲン原子、R1はエチニル基又はシアノ基、R2は水素を示す。)
【0048】
Pで表される5’位水酸基の保護基としては、1級水酸基の選択的な保護基として常用されているものであればよく、具体的には、トリメトキシトリチル基、ジメトキシトリチル基、メトキシトリチル基、トリチル基、t−ブチルジメチルシリル基、t−ブチルジフェニルシリル基、ベンゾイル基等を例示することができる。
【0049】
保護基の導入反応は、前記と同様の方法にて実施できる。
【0050】
第2工程;
第2工程は、式[VIII]で表される化合物の3’位水酸基を脱水し、2’,3’−2重結合とし、化合物式[VIV]とする工程である。
【0051】
【化7】
【0052】
(式中、Pは保護基、Xはハロゲン原子、R1はエチニル基又はシアノ基を示す。)
化合物[VIII]の3’位水酸基を脱水し、化合物[VIV]とするには、化合物[VIII]の3’位水酸基をハロゲン原子、又は、メタンスルホン酸、クロロメタンスルホン酸、トルエンスルホン酸、トリフルオロメタンスルホン酸等のスルホン酸エステルのような脱離能を有する官能基へと変換後、これを塩基処理により脱離させることで実施できる。
【0053】
反応条件は用いる試薬により異なり、たとえば、トリフルオロメタンスルホン酸エステルを経る反応を用いる場合、ジクロロメタン、ピリジン等の有機溶媒中、トリフルオロメタンスルホン酸無水物とピリジン、トリエチルアミン等の塩基をそれぞれ、1〜5モル、5〜10モル用い、−78℃〜室温で1時間〜24時間程度反応させることにより実施できる。
【0054】
こうして得られた化合物[VIV]の保護基を除去して、R2が水素である本発明化合
物を得、必要に応じてリン酸化して本発明化合物を得る。
【0055】
【化8】
【0056】
(式中、Xはハロゲン原子、Pは保護基、R1はエチニル基又はシアノ基、R2は、水素原子、リン酸又はその誘導体残基を示す。)
【0057】
保護基の除去は、使用した保護基に応じ、酸性加水分解、アルカリ性加水分解、フッ化テトラブチルアンモニウム処理、接触還元などの通常の処理方法から適宜選択して行えばよい。
【0058】
R2がリン酸又はその誘導体残基を示す化合物は、前記式[I]化合物と同様の方法で合成することができる。
【0059】
本発明化合物[III]は、以下に説明する工程により製造することができる。
第1工程;
第1工程は、式[X]で表される化合物の4位のヒドロキシメチル基を酸化してアルデヒド化合物とし、更にトリエチルシリルエチニル化合物、又はシアノ化合物へと変換することにより式[XI]で表される化合物を得る工程である。
【0060】
【化9】
【0061】
(R1はエチニル基、トリエチルシリルエチニル基又はシアノ基を示す。)
【0062】
原料化合物は、式[X]で表される公知化合物である(Biosci.Biotech.Biochem.,57,1433−1438(1993))。トリエチルシリルエチニル化合物への変換反応は、式[X]化合物の4位ヒドロキシメチル基をホルミル基へと酸化した後、これをジブロモビニル基へと変換し、引き続き、強塩基処理するにより脱ブロモ化水素することにより実施できる。
【0063】
式[X]で表される化合物の4−ヒドロキシメチル基をホルミル基に変換する場合、用いる酸化剤としては、無水クロム酸、ピリジンと無水酢酸との複合試薬、ピリジンクロロクロメート、ピリジンジクロメートなどのクロム系酸化剤;デス−マーチン試薬などの高原子価ヨウ素酸化剤;ジメチルスルホキシドと無水酢酸、塩化オキサリル又はジシクロヘキシルカルボジイミドとを組み合わせて用いるジメチルスルホキシド系酸化剤などを例示することができる。
【0064】
反応条件は用いる酸化剤により異なり、たとえば、塩化オキサリルとジメチルスルホキシドを用いて酸化する場合、ジクロロメタンなどの有機溶媒中、必要によりアルゴン、窒素などの不活性ガス雰囲気下、式[X]化合物1モルに対して塩化オキサリルとジメチルスルホキシドをそれぞれ1〜5モル、1.5〜6モル用い、−100〜0℃で15分から2時間程度反応させ、トリエチルアミンなどの塩基類を2〜10モル添加し、更に室温にて15分から2時間程度反応させることにより実施できる。
【0065】
次に、得られたアルデヒド化合物からアルキン化合物への変換は、アルデヒド化合物を増炭反応(C−C結合形成反応)に付し、強塩基で処理してメタルアルキニル化合物とし、最後に保護基を導入することにより実施できる。増炭反応は、ジクロロメタン、ジクロロエタンなどの有機溶媒中、必要によりアルゴン、窒素などの不活性ガス雰囲気下、先に得られたアルデヒド化合物1モルに対して四臭化炭素とトリフェニルホスフィンをそれぞれ1〜5モルと2〜10モル用い、0〜50℃で15分〜3時間程度反応させることにより実施できる。
【0066】
強塩基処理は、テトラヒドロフラン、1,4−ジオキサン、ジメトキシエタンなどの有機溶媒中、必要によりアルゴン、窒素などの不活性ガス雰囲気下、増炭反応により得られた化合物1モルに対してメチルリチウム、n−ブチルリチウム、t−ブチルリチウムなどのリチウム化合物2〜4モル用い、−100〜−20℃で5〜60分程度反応させることにより実施できる。更に、得られた化合物のアルキニル基にシリル系保護基を導入する場合、上記強塩基処理に続けてクロロトリエチルシランなどのシリル化剤を添加し、反応させることにより実施することができる。
【0067】
一方、式[X]化合物のシアノ化合物への変換反応は、式[X]化合物の4位ヒドロキシメチル基をホルミル基へと酸化した後、これをオキシム化合物へと変換し、引き続き、得られたオキシム化合物を脱水することにより実施できる。
【0068】
式[X]で表される化合物の4−ヒドロキシメチル基をホルミル基に変換する場合、用いる酸化剤としては、無水クロム酸、ピリジンと無水酢酸との複合試薬、ピリジンクロロクロメート、ピリジンジクロメートなどのクロム系酸化剤;デス−マーチン試薬などの高原子価ヨウ素酸化剤;ジメチルスルホキシドと無水酢酸、塩化オキサリル又はジシクロヘキシルカルボジイミドとを組み合わせて用いるジメチルスルホキシド系酸化剤などを例示することができる。
【0069】
反応条件は用いる酸化剤により異なり、たとえば、塩化オキサリルとジメチルスルホキシドを用いて酸化する場合、ジクロロメタンなどの有機溶媒中、必要によりアルゴン、窒素などの不活性ガス雰囲気下、式[X]化合物1モルに対して塩化オキサリルとジメチルスルホキシドをそれぞれ1〜5モル、1.5〜6モル用い、−100〜0℃で15分から2時間程度反応させ、トリエチルアミンなどの塩基類を2〜10モル添加し、更に室温にて15分から2時間程度反応させることにより実施できる。
【0070】
アルデヒド化合物のオキシム化合物への変換は、ピリジン等の有機溶媒中、アルデヒド化合物1モルに対して1〜5モルの塩酸ヒドロキシルアミンを使用し、室温〜100℃で30分〜3時間程度反応させることにより実施できる。
【0071】
オキシム化合物の脱水は、ジクロロメタン、アセトニトリル、テトラヒドロフラン等の有機溶媒中、ピリジン、トリエチルアミン、酢酸ナトリウム等の塩基存在下、ホスゲン、カルボニルジイミダゾール、塩化メタンスルホニル、無水酢酸等の脱水剤を用いることで実施できる。
【0072】
脱水反応条件は用いる脱水剤により異なり、たとえば、塩化メタンスルホニルを用いて脱水する場合、ジクロロメタン、テトラヒドロフラン、ピリジン等の有機溶媒中、オキシム化合物1モルに対して塩化メタンスルホニルとトリエチルアミンをそれぞれ1〜5モル、5〜10モル用い、−50℃〜室温で15分〜2時間程度反応させることにより実施できる。
【0073】
第2工程;
第2工程は、式[XI]で表される化合物の3,5位の水酸基を保護するメトキシベンジリデン基を除去し、式[XII]で表される化合物とする工程である。
【0074】
【化10】
【0075】
(R1はエチニル基、トリエチルシリルエチニル基又はシアノ基を示す。)
【0076】
保護基の除去法としては、酸性加水分解、接触還元等の処理方法から適宜選択して行えばよい。
【0077】
反応条件は用いる反応により異なり、たとえば、酸性加水分解を用いるときは、式[XI]の化合物をギ酸、酢酸等の有機酸、又は鉱酸の水溶液中、0〜100℃で1〜24時間反応させることにより実施できる。
【0078】
第3工程;
第3工程は式[XII]で表される化合物の5位水酸基を選択的に保護し、式[XIII]で表される化合物を得る工程である。
【0079】
【化11】
【0080】
(式中、Pは保護基、R1はエチニル基、トリエチルシリルエチニル基又はシアノ基を示す。)
【0081】
Pで表される5位水酸基の保護基としては、1級水酸基の選択的な保護基として常用されているものであればよく、具体的には、トリメトキシトリチル基、ジメトキシトリチル基、メトキシトリチル基、トリチル基、t−ブチルジメチルシリル基、t−ブチルジフェニルシリル基、ベンゾイル基等を例示することができる。
保護基の導入反応は、前記と同様の方法にて実施できる。
【0082】
第4工程;
第4工程は式[XIII]で表される化合物の3位水酸基を還元し、式[XIV]で表される化合物を得る工程である。
【0083】
【化12】
【0084】
(式中、Pは保護基、R1はエチニル基、トリエチルシリルエチニル基又はシアノ基を示す。)
【0085】
3位水酸基のデオキシ化は、3位水酸基をハロゲン化体(ヨウ素体、臭素体、塩素体)、フェノキシチオカルボニル体、チオカルボニルイミダゾール体、メチルジチオカルボネート体等に変換した後、ラジカル開始剤存在下、ラジカル還元剤により還元することによって行うことができる。
【0086】
例えば、フェノキシチオカルボニル体に導いてデオキシ化する場合、フェノキシチオカルボニル化反応は、必要によりアルゴン、窒素等の不活性ガス雰囲気下、テトラヒドロフラン、アセトニトリル、ジクロロメタン等の有機溶媒中、ジメチルアミノピリジン、ピリジン等の塩基共存下、3位水酸基の保護基のみ除去された上記化合物1モルに対してクロロチオノギ酸フェニル誘導体1〜10モル、好ましくは1〜2モル用い、0〜50℃で0.5〜5時間程度撹拌反応させることにより実施することができる。
【0087】
続けて行う還元反応は、トルエン、ベンゼン等の有機溶媒中、必要によりアルゴン、窒素等の不活性ガス雰囲気下、アゾビスイソブチロニトリル等のラジカル開始剤存在下、上記フェノキシチオカルボニル体1モルに対して水素化トリブチルスズ、トリス(トリメチルシリル)シラン等のラジカル還元剤1〜10モル、好ましくは2〜5モル用い、50〜150℃で1〜5時間程度撹拌反応させることにより実施することができる。
【0088】
第5工程;
第5工程は式[XIV]で表される化合物の1,2位イソプロピリデン基を除去後、生じた水酸基をアセチル化し、式[XV]で表される化合物を得る工程である。
【0089】
【化13】
【0090】
(式中、Pは保護基、R1はエチニル基、トリエチルシリルエチニル基又はシアノ基を示す。)
【0091】
1,2位イソプロピリデン基の除去法として、酸性加水分解の条件を用いるときは、式[XIV]化合物をギ酸、酢酸等の有機酸、又は鉱酸の水溶液中、0〜100℃で1〜24時間反応させることにより実施できる。
【0092】
引き続く、水酸基へのアセチル基の導入は、常法により行うことができ、たとえば、ピリジン、アセトニトリル、ジクロロメタン等の有機溶媒中、ピリジン、トリエチルアミン等の塩基存在下、塩化アセチル、無水酢酸などのアセチル化剤と反応させることにより実施することができる。
【0093】
たとえば、ピリジン中、無水酢酸により反応を行う場合、上記の脱イソプロピリデン体1モルに対して、2〜10モルの無水酢酸、及び、場合に応じて、触媒量の4−ジメチルアミノピリジンを用い、0〜100℃で1〜24時間反応させることにより実施できる。
【0094】
第6工程;
第6工程は式[XV]で表される化合物を、2,6−ジアミノプリンと縮合し、式[XVI]で表される化合物を得る工程である。
【0095】
【化14】
【0096】
(式中、Pは保護基、R1はエチニル基、トリエチルシリルエチニル基又はシアノ基を示す。)
【0097】
式[XV]で表される化合物と2,6−ジアミノプリンとの縮合は、ルイス酸存在下、式[XV]の化合物を2,6−ジアミノプリンと反応させることによって行うことができる。2,6−ジアミノプリンはシリル化したものを用いてもよく、このようなシリル化した塩基類は公知の方法、たとえばヘキサメチルジシラザンとトリメチルクロロシラン中で加熱環流するか、又はアセトニトリル、1,2−ジクロロエタン等の有機溶媒中、ビス(トリメチルシリル)アセトアミドと加熱還流することにより得ることができる。使用するルイス酸としては、トリフルオロメタンスルホン酸トリメチルシリル、四塩化すず、塩化亜鉛、ヨウ化亜鉛、無水塩化アルミニウムなどが例示される。
【0098】
縮合反応は、ジクロロメタン、1,2−ジクロロエタン、アセトニトリル、トルエン等の有機溶媒中、必要によりアルゴン、窒素などの不活性ガス雰囲気下、式[XV]の化合物1モルに対し2,6−ジアミノプリン1〜10モル及びルイス酸0.1〜10モルとを用い、−20〜150℃で30分〜24時間程度反応させることにより実施することができる。
【0099】
第7工程;
第7工程は、式[XVI]で表される化合物の2位アミノ基をフッ素又は塩素へと変換し、式[XVII]で表される化合物を得る工程である。
【0100】
【化15】
【0101】
(式中、Pは保護基、Xはハロゲン原子、R1はエチニル基、トリエチルシリルエチニル基又はシアノ基を示す。)
【0102】
式[XVII]で表される化合物は、式[XVI]で表される化合物の2位アミノ基を亜硝酸誘導体で処理後、ハロゲン化試薬を用いて塩基部2位にハロゲン原子を導入するか、又は、2,6位のアミノ基を同条件下処理し、2,6−ジハロプリン誘導体とした後、アンモニア処理により塩基部6位のハロゲン原子をアミノ基とすることで合成することができる。
【0103】
式[XVI]で表される化合物の2位アミノ基をフッ素へと置換する試薬としては、テトラフルオロホウ酸中の亜硝酸ナトリウム、フッ化水素−ピリジン中の亜硝酸t−ブチル等の亜硝酸エステルなどを例示することができる。
【0104】
反応条件は用いる試薬により異なり、たとえば、フッ化水素−ピリジン中、亜硝酸t−ブチルを用いる場合、フッ化水素−ピリジンを溶媒として、亜硝酸t−ブチルを1〜3モル用い、−50℃から0℃で15分から5時間程度反応させればよい。また、2,6−ジフルオロプリン誘導体とした場合は、引き続き、ジオキサン、メタノール等の有機溶媒中、アンモニア水処理すればよい。
【0105】
一方、式[XVI]で表される化合物の2位アミノ基を塩素へと置換する試薬としては、ジクロロメタンなどの有機溶媒中、三塩化アンチモン−亜硝酸t−ブチル、塩化アセチル−亜硝酸ベンジルトリエチルアンモニウムの組み合わせなどを例示することができる。
【0106】
反応条件は用いる試薬により異なり、たとえば、塩化アセチル−亜硝酸ベンジルトリエチルアンモニウムの組み合わせを用いる場合、ジクロロメタン等の有機溶媒中、化合物[XVI]1モルに対して、亜硝酸ベンジルトリエチルアンモニウム1〜5モルを−50℃から室温で30分間から3時間程度、1〜5モルの塩化アセチルと処理した後、化合物[XVI]を−50℃から室温で1時間から数日間反応させればよい。また、2,6−ジクロロプリン誘導体とした場合は、引き続き、ジオキサン、メタノール等の有機溶媒中、アンモニア水処理すればよい。
【0107】
第8工程;
第8工程は、式[XVII]で表される化合物の2’位水酸基を保護するアセチル基を除去し、式[XVIII]で表される化合物を得る工程である。
【0108】
【化16】
【0109】
(式中、Pは保護基、Xはハロゲン原子、R1はエチニル基、トリエチルシリルエチニル基又はシアノ基を示す。)
【0110】
アセチル基の除去は、適当な塩基又は酸触媒を用いて行うことができ、例えば塩基触媒を用いる場合、エタノール等のアルコール系溶媒と水との混合溶媒中、水酸化ナトリウム、水酸化カリウム、トリエチルアミン、アンモニア水等を例示することができる。
【0111】
例えば、メタノール中、アンモニア水を用い、0〜100℃で1〜24時間反応させることにより、実施できる。
【0112】
第9工程;
第9工程は、式[XVIII]で表される化合物の2’位水酸基を還元し、式[XIX]で表される化合物を得る工程である。
【0113】
【化17】
【0114】
(式中、Pは保護基、Xはハロゲン原子、R1はエチニル基、トリエチルシリルエチニル基又はシアノ基を示す。)
【0115】
2’位水酸基のデオキシ化は、2’位水酸基をハロゲン化体(ヨウ素体、臭素体、塩素体)、フェノキシチオカルボニル体、チオカルボニルイミダゾール体、メチルジチオカルボネート体等に変換した後、ラジカル開始剤存在下、ラジカル還元剤により還元することによって行うことができる。
【0116】
例えば、フェノキシチオカルボニル体に導いてデオキシ化する場合、フェノキシチオカルボニル化反応は、必要によりアルゴン、窒素等の不活性ガス雰囲気下、テトラヒドロフラン、アセトニトリル、ジクロロメタン等の有機溶媒中、ジメチルアミノピリジン、ピリジン等の塩基共存下、2’位水酸基の保護基のみ除去された上記化合物1モルに対してクロロチオノギ酸フェニル誘導体1〜10モル、好ましくは1〜2モル用い、0〜50℃で0.5〜5時間程度撹拌反応させることにより実施することができる。
【0117】
続けて行う還元反応は、トルエン、ベンゼン等の有機溶媒中、必要によりアルゴン、窒素等の不活性ガス雰囲気下、アゾビスイソブチロニトリル等のラジカル開始剤存在下、上記フェノキシチオカルボニル体1モルに対して水素化トリブチルスズ、トリス(トリメチルシリル)シラン等のラジカル還元剤1〜10モル、好ましくは2〜5モル用い、50〜150℃で1〜5時間程度撹拌反応させることにより実施することができる。
【0118】
こうして得られた化合物[XIX]の水酸基、及び、エチニル基の保護基を除去して、R2が水素である本発明化合物を得、必要に応じてリン酸化して本発明化合物を得る。
【0119】
【化18】
【0120】
(式中、Pは保護基、Xはハロゲン原子、R1はエチニル基、トリエチルシリルエチニル基又はシアノ基、R2は、水素、リン酸又はその誘導体残基を示す。)
【0121】
保護基の除去は、使用した保護基に応じ、酸性加水分解、アルカリ性加水分解、フッ化テトラブチルアンモニウム処理、接触還元などの通常の処理方法から適宜選択して行えばよい。
また、R2がリン酸又はその誘導体残基を示す化合物は、前記式[I]化合物と同様の方法で合成することができる。
【0122】
本発明化合物は、一般のヌクレオシド、ヌクレオチドの単離精製に使用されている方法(例えば、再結晶法、イオン交換カラムクロマトグラフィー、吸着カラムクロマトグラフィーなど)を適宜組み合せて分離精製することができる。このようにして得られた化合物は、必要に応じて塩とすることもできる。
【0123】
(3)用途
本発明化合物は、後述の試験例に示すようにレトロウイルスに対して優れた抗ウイルス作用を有することから、これらを有効成分とする本発明組成物は医薬としての使用、具体的にはレトロウイルスの感染症の処置、特にヒト免疫不全ウイルス(HIV)感染に起因するエイズの治療に有用である。
【0124】
本発明化合物の投与量は、患者の年齢、体重、疾病、患者の重篤度、薬物による忍容性、投与方法などにより異なり、これらの条件を総合した上で適宜決定されるものであるが、通常1日当たり0.00001〜1000mg/kg体重、好ましくは0.0001〜100mg/kg体重の範囲内から選ばれ、一回又は複数回に分けて投与される。
投与方法は、経口、非経口、経腸、局所投与などのいずれの経路でもよい。
【0125】
本発明化合物の製剤化に際しては、通常使用される製剤用担体、賦形剤、その他の添加剤を含む組成物として使用するのが普通である。担体としては、乳糖、カオリン、ショ糖、結晶セルロース、コーンスターチ、タルク、寒天、ペクチン、ステアリン酸、ステアリン酸マグネシウム、レシチン、塩化ナトリウムなどの固体状担体、グリセリン、落花生油、ポリビニルピロリドン、オリーブ油、エタノール、ベンジルアルコール、プロピレングリコール、水などの液状担体を例示することができる。
【0126】
剤型としては任意の形態を採ることができ、たとえば固体状担体を使用する場合には錠剤、散剤、顆粒剤、カプセル化剤、坐剤、トローチ剤などを、液状担体を使用する場合にはシロップ、乳液、軟ゼラチンカプセル、クリーム、ゲル、ペースト、スプレー、注射などをそれぞれ例示することができる。
【実施例】
【0127】
以下、本発明を合成例、試験例、製剤例などをあげて具体的に説明するが、本発明はこれらによって何等限定されるものではない。
【0128】
合成例1:2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン(化合物4)の合成
【0129】
(1)9−(3,5−ジ−O−アセチル−2−デオキシ−4−C−エチニル−β−D−リボ−ペントフラノシル)−2,6−ジアミノプリン(化合物2)の合成
【0130】
【化19】
【0131】
化合物1(0.33g、1.14mmol)をアセトニトリル(10.0mL)に懸濁し、無水酢酸(0.23mL、2.43mmol)、トリエチルアミン(0.67g、4.81mmol)、4−ジメチルアミノピリジン少量を加え、室温で終夜撹拌した。
析出した結晶をろ取、乾燥し、化合物2(0.40g、1.07mmol、93.9%)を得た。
【0132】
1H−NMR(DMSO−d6)δ:7.94(1H,s,H−8),6.76(2H,bs,NH2),6.27(1H,t,H−1’,J=7.00),5.84(2H,bs,NH2),5.60(1H,dd,H−3’,J=4.00,6.80),4.46(1H,d,H−5’a,J=11.5),4.21(1H,d,H−5’b,J=11.5),3.74(1H,s,ethynyl)3.12(1H,m,H−2’a),2.52(1H,m,H−2’b),2.12,2.03(each3H,s,acetyl)
【0133】
(2)3’,5’−ジ−O−アセチル−2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン(化合物3)の合成
【0134】
【化20】
【0135】
化合物2(450mg、1.20mmol)を70%フッ化水素−ピリジン(5.00mL)に溶解し、亜硝酸t−ブチル(0.194mL、1.63mmol)を加え、−10℃で1時間撹拌した。蒸留水を加えた後、クロロホルムで抽出し、有機層を無水硫酸マグネシウム上で乾燥後、減圧下濃縮した。残さにクロロホルム:メタノール(50:1)を加え、析出した結晶をろ取、乾燥し、化合物3(240mg、0.64mmol、53.3%)を得た。
【0136】
1H−NMR(DMSO−d6)δ:8.34(1H,s,H−8),7.94,7.99(each1H,bs,NH2),6.35(1H,t,H−1’,J=6.80),5.68(1H,dd,H−3’,J=5.10,7.05),4.41(1H,d,H−5’a,J=11.6),4.21(1H,d,H−5’b,J=11.6),3.42(1H,s,ethynyl),3.14(1H,m,H−2’a),2.63(1H,m,H−2’b),2.12,2.00(each3H,s,acetyl).
【0137】
(3)2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン(化合物4)の合成
【0138】
【化21】
【0139】
化合物3(200mg、0.53mmol)をメタノール(7.00mL)に溶解し、28%アンモニア水(5.00mL)を加え、室温で4時間撹拌した。反応液を減圧下濃縮後、残さにクロロホルム:メタノール(20:1)を加え、析出した結晶をろ取した。得られた結晶を水から再結晶化し、化合物4(113mg、0.39mmol、73.6%)を得た。
【0140】
1H−NMR(DMSO−d6)δ:8.30(1H,s,H−8),7.87,7.84(each1H,bs,NH2),6.24(1H,dd,H−1’,J=5.05,7.15),5.57(1H,d,3’−OH,J=5.50),5.30(1H,t,5’−OH,J=6.40),4.57(1H,m,H−3’),3.65(1H,m,H−5’a),3.55(1H,m,H−5’b),3.51(1H,s,ethynyl),2.70(1H,m,H−2’a),2.44(1H,m,H−2’b).
【0141】
合成例2:4’−C−シアノ−2’−デオキシ−2−フルオロアデノシン(化合物8)の合成
【0142】
(1)9−(3,5−ジ−O−アセチル−4−C−シアノ−2−デオキシ−β−D−リボ−ペントフラノシル)−2,6−ジアミノプリン(化合物6)の合成
【0143】
【化22】
【0144】
化合物5(122mg、0.418mmol)をアセトニトリル(5.00mL)に懸濁し、無水酢酸(118μl、1.25mmol)、トリエチルアミン(352μl、2.51mmol)、4−ジメチルアミノピリジン少量を加え、室温で終夜撹拌した。析出した結晶をろ取、乾燥し、化合物6(128mg、0.341mmol、81.6%)を得た。
【0145】
1H−NMR(CDCl3)δ:7.54(1H,s,H−8),6.31(1H,t,H−1’,J=7.00),6.06(1H,dd,H−3’,J=5.00,6.50),5.31(2H,bs,NH2),4.95(1H,d,H−5’a,J=11.5),4.80(2H,bs,NH2),4.37(1H,d,H−5’b,J=12.0),3.43(1H,m,H−2’a),2.63(1H,m,H−2’b),2.23,2.12(each3H,s,acetyl).
【0146】
(2)3’,5’−ジ−O−アセチル−4’−C−シアノ−2’−デオキシ−2−フルオロアデノシン(化合物7)の合成
【0147】
【化23】
【0148】
化合物6(118mg、0.314mmol)を70%フッ化水素−ピリジン(2.30mL)に溶解し、亜硝酸t−ブチル(50.0μl、0.427mmol)を加え、−10℃で3時間撹拌した。亜硝酸t−ブチル(10.0μl、85μmol)を追加し、同温度で更に1時間撹拌した。飽和重曹水を加えた後、酢酸エチルで抽出し、有機層を飽和食塩水で洗浄した。有機層を硫酸マグネシウム上で乾燥後、減圧濃縮した。残さをエタノールに加熱溶解後、冷却し、析出した結晶をろ取、乾燥し、化合物7(53.7mg、0.14mmol、45.2%)を得た。
【0149】
1H−NMR(DMSO−d6)δ:8.35(1H,s,H−8),8.00,7.92(each1H,bs,NH2),6.54(1H,t,H−1’,J=7.00),5.83(1H,dd,H−3’,J=4.00,6.50),4.63(1H,d,H−5’a,J=11.5),4.44(1H,d,H−5’b,J=12.0),3.26(1H,m,H−2’a),2.73(1H,m,H−2’b),2.18,2.05(each3H,s,acetyl).
【0150】
(3)4’−C−シアノ−2’−デオキシ−2−フルオロアデノシン(化合物8)の合成
【0151】
【化24】
【0152】
化合物7(53.7mg、0.142mmol)をメタノール(1.90mL)に溶解し、28%アンモニア水(1.30mL)を加え、室温で30分間撹拌した。反応液を減圧下濃縮後、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 10mL、ヘキサン:酢酸エチル=5:1〜酢酸エチル〜酢酸エチル:メタノール=10:1)で精製し、化合物8(30.2mg、0.10mmol、72.3%)を得た。
【0153】
1H−NMR(DMSO−d6)δ:8.31(1H,s,H−8),7.93,7.82(each1H,bs,NH2),6.43(1H,t,H−1’,J=7.00),6.36(1H,bs,3’−OH),5.74(1H,bs,5’−OH),4.70(1H,t,H−3’,J=5.50),3.80(1H,d,H−5’a,J=12.0),3.65(1H,d,H−5’b,J=12.0),2.93(1H,m,H−2’a),2.47(1H,m,H−2’b).
【0154】
合成例3:2−クロロ−2’−デオキシ−4’−C−エチニルアデノシン(化合物9)の合成
【0155】
【化25】
【0156】
亜硝酸ベンジルトリエチルアンモニウム(1.04g、4.36mmol)をジクロロメタン(24.0mL)に溶解し、塩化アセチル(0.40mL、5.63mmol)を加え、0℃で30分間撹拌した。この溶液に、化合物2(340mg、0.91mmol)のジクロロメタン(6.00mL)溶液を加え、0℃で3時間撹拌した。反応液をクロロホルムで希釈した後、有機層を水洗し、無水硫酸マグネシウム上で乾燥、減圧下濃縮した。残さに28%アンモニア水(10.0mL)、メタノール(15.0mL)を加え、室温で終夜撹拌した後、反応液を減圧下濃縮し、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 50mL、クロロホルム:メタノール=20:1〜10:1)で精製した。得られた残さを更にODSカラムクロマトグラフィー(ODS 50mL、5〜10〜15〜20%アセトニトリル)により精製し、化合物9(39.2mg、0.13mmol、14.3%)を得た。
【0157】
1H−NMR(DMSO−d6)δ:8.34(1H,s,H−8),7.84(2H,bs,NH2),6.27(1H,dd,H−1’,J=5.00,7.00),5.60(1H,d,3’−OH,J=5.00),5.33(1H,t,5’−OH,J=6.00),4.56(1H,m,H−3’),3.64(1H,m,H−5’a),3.56(1H,m,H−5’b),3.52(1H,s,ethynyl),2.68(1H,m,H−2’a),2.45(1H,m,H−2’b).
【0158】
合成例4:2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン5’−H−ホスホネート(化合物10)の合成
【0159】
【化26】
【0160】
化合物4(50.0mg、0.171mmol)をピリジン(2.00mL)に溶解し、ホスホン酸(21.0mg,0.25mmol)、ジシクロヘキシルカルボジイミド(106mg、0.51mmol)を加え、室温で5時間撹拌した。沈殿をろ去後、ろ液を減圧下濃縮し、残さを水−クロロホルムで分配した。水層を減圧下濃縮し、残さを分取薄層クロマトグラフィーにより精製した(イソプロパノール:28%アンモニア水:水=7:1:2)。得られた残さをアセトニトリルで共沸後、メタノール−エーテルで粉末とし、化合物10(6.3mg、17.6μmol、10.3%)を得た。
【0161】
1H−NMR(D2O)δ:8.13(1H,s,H−8),6.49(1H,d,H−P,J=645),6.25(1H,dd,H−1’,J=5.00,7.50),3.96(2H,m,H−5’),2.75,2.59(each 1H,m,H−2’).
【0162】
31P−NMR(D2O)δ:6.45.
【0163】
合成例5:2’,3’−ジデヒドロ−2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン(化合物13)の合成
【0164】
【化27】
【0165】
化合物4(0.28g、0.95mmol)をジメチルホルムアミド(7.00mL)に溶解し、t−ブチルクロロジフェニルシラン(0.50mL,1.92mmol)、イミダゾール(0.26g、3.82mmol)を加え、室温で終夜撹拌した。反応液にメタノールを加えた後、減圧下濃縮し、残さを酢酸エチル−水で分配した。有機層を無水硫酸マグネシウム上で乾燥後、減圧下濃縮し、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 100mL、クロロホルム:メタノール=20:1)で精製した。粗製の化合物11(0.38g)を得た。
粗製の化合物11(0.38g)をジクロロメタン(10.0mL)に溶解し、−10℃で無水トリフルオロメタンスルホン酸(0.14mL,0.83mmol)、ピリジン(0.14g、1.71mmol)を加え、同温度で2時間撹拌した。反応液に飽和重曹水を加えた後、クロロホルムで抽出した。有機層を無水硫酸マグネシウム上で乾燥後、減圧下濃縮し、得られた粗トリフラートを精製することなく、次の反応に用いた。
【0166】
粗トリフラートを乾燥テトラヒドロフラン(20.0mL)に溶解し、アルゴン雰囲気下、−78℃で、1M ナトリウムヘキサメチルジシラジド−テトラヒドロフラン溶液(2.50mL、2.50mmol)を加え、同温度で2時間撹拌後、反応液を室温に戻し、終夜撹拌した。反応液に水を加えた後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウム上で乾燥後、減圧下濃縮し、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 50mL、クロロホルム:メタノール=50:1〜20:1)で精製し、粗製の化合物12(0.20g)を得た。
得られた粗製の化合物12をテトラヒドロフラン(10.0mL)に溶解し、1M フッ化テトラブチルアンモニウム−テトラヒドロフラン溶液(0.59mL、0.59mmol)を加え、室温で30分間撹拌した。反応液を減圧下濃縮後、クロロホルム:メタノール(10:1)を加え、析出した結晶をろ取した。化合物13(52.0mg、0.19mmol、20.0% from 化合物4)を得た。
【0167】
1H−NMR(DMSO−d6)δ:8.08(1H,s,H−8),7.84(2H,bs,NH2),6.90(1H,t,H−1’,J=1.50),6.43(1H,dd,H−3’,J=2.00,6.00),6.27(1H,dd,H−3’,J=1.00,6.00),5.37(1H,t,5’−OH,J=6.00)3.71(1H,s,ethynyl),3.67(1H,dd,H−5’a,J=6.00,12.0),3.57(1H,dd,H−5’b,J=6.00,12.0).
【0168】
合成例6:2’,3’−ジデヒドロ−2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン5’−H−ホスホネート(化合物14)の合成
【0169】
【化28】
【0170】
化合物13(13.0mg、0.047mmol)をピリジン(0.7mL)に溶解し、ホスホン酸(7.7mg,0.094mmol)、ジシクロヘキシルカルボジイミド(29.2mg、0.14mmol)を加え、室温で1時間撹拌した。反応液を減圧下濃縮し、残さをODSカラムクロマトグラフィー(ODS 10mL、0〜1%アセトニトリル)により精製した。得られた残さをDowex 50Wx8カラム(Na型)に通過させた。通過液を濃縮してえられた残さをメタノール−エーテルで粉末とし、化合物14(4.3mg、12μmol、25.5%)を得た。
【0171】
1H−NMR(MeOD)δ:8.30(1H,s,H−8),6.69(1H,d,H−P,J=625),7.02(1H,bt,H−1’),6.48(1H,dd,H−2’,J=2.00,5.50),6.22(1H,dd,H−3’,J=1.00,5.50),4.18(1H,dd,H−5’a,J=7.50,11.0),3.99(1H,dd,H−5’b,J=7.50,11.0).
【0172】
31P−NMR(MeOD)δ:4.11.
【0173】
合成例7:2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン(化合物23)の合成
【0174】
(1)1,2−O−イソプロピリデン−4−C−トリエチルシリルエチニル−α−D−キシロ−ペンタフラノース(化合物16)の合成
【0175】
【化29】
【0176】
塩化オキサリル(0.54mL、6.19mmol)をジクロロメタン(10.0mL)に溶解後、−60℃でジメチルスルホキシド(0.90mL、12.7mmol)を滴下し、同温度で15分間撹拌した。ここに化合物15(1.06g、3.13mmol、Biosci.Biotech.Biochem.,57,1433−1438(1993))のジクロロメタン溶液(15.0mL)を滴下し、同温度で30分間撹拌した。トリエチルアミン(1.86mL、13.3mmol)を加えた後、反応液を室温に戻し、30分間撹拌した。反応液をクロロホルムで希釈後、水洗し、有機層を無水硫酸マグネシウム上で乾燥した。有機層を減圧下濃縮し、得られた粗アルデヒドを精製することなく次の反応に用いた。
粗アルデヒドをジクロロメタン(40.0mL)に溶解し、0℃で四臭化炭素(2.08g、6.27mmol)、トリフェニルホスフィン(3.28g、12.5mmol)を加えた後、室温で1時間撹拌した。反応液にトリエチルアミン(2.60mL、18.7mmol)を加えた後、クロロホルムで希釈し、有機層を水洗した。有機層を無水硫酸マグネシウム上で乾燥後、減圧下濃縮し、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 100mL、ヘキサン:酢酸エチル=3:1)で精製した。粗ジブロモエテン(1.42g)を得た。
【0177】
粗ジブロモエテン(1.42g、2.89mmol)を乾燥テトラヒドロフラン(20.0mL)に溶解し、アルゴン雰囲気下、−10℃で2.2M メチルリチウム−エーテル溶液(4.49mL、9.88mmol)を加え、同温度で5分間撹拌した。ここにクロロトリエチルシラン(0.95mL、5.66mmol)を加え、更に30分間撹拌した。反応液に飽和塩化アンモニウム水溶液を加え撹拌後、酢酸エチルで抽出した。有機層を無水硫酸マグネシウム上で乾燥後、減圧下濃縮し、得られた粗アルキンを得た。
粗アルキンを酢酸(80.0mL)に溶解し、水(20.0mL)を加え、室温で終夜撹拌した。反応液を減圧下濃縮し、得られた残さをトルエンで共沸した。残さをシリカゲルカラムクロマトグラフィー(シリカゲル 50mL、ヘキサン:酢酸エチル=3:1)で精製し、化合物16(0.70g、2.13mmol、64.4%)を得た。
【0178】
1H−NMR(CDCl3)δ:6.00(1H,d,H−1,J=3.50),4.60(1H,d,H−2,J=4.00),4.58(1H,d,H−3,J=5.00),3.96−3.91(3H,m,H−5 and 3−OH),2.50(1H,t,5−OH),1.64,1.33(each3H,s,acetonide),0.97(9H,t,Et,J=8.00),0.59(6H,q,Et,J=8.00).
【0179】
(2)5−O−t−ブチルジフェニルシリル−1,2−O−イソプロピリデン−4−C−トリエチルシリルエチニル−α−D−キシロ−ペンタフラノース(化合物17)の合成
【0180】
【化30】
【0181】
化合物16(0.70g、2.13mmol)をジメチルホルムアミド(3.50mL)に溶解し、t−ブチルクロロジフェニルシラン(0.66mL、2.54mmol)、イミダゾール(0.35g、5.14mmol)を加え、終夜撹拌した。反応液にメタノールを加え減圧下濃縮し後、残さを酢酸エチルに溶解した。有機層を水洗後、無水硫酸マグネシウム上で乾燥し、減圧下濃縮した。残さをシリカゲルカラムクロマトグラフィー(シリカゲル 100mL、ヘキサン:酢酸エチル=5:1)で精製し、化合物17(1.10g、1.94mmol、91.1%)を得た。
【0182】
1H−NMR(CDCl3)δ:7.72−7.36(10H,s,aromatic),6.02(1H,d,H−1,J=3.50),4.66(1H,d,H−3,J=5.50),4.63(1H,d,H−1,J=4.00),4.05(1H,d,H−5a,J=10.5),3.99(1H,d,3−OH,J=5.50),3.93(1H,d,H−5’b,J=10.5),1.65,1.35(each3H,s,acetonide),1.06(9H,s,t−Bu),0.93(9H,t,Et,J=8.00),0.56(6H,q,Et,J=8.00).
【0183】
(3)5−O−t−ブチルジフェニルシリル−3−デオキシ−1,2−O−イソプロピリデン−4−C−トリエチルシリルエチニル−α−D−キシロ−ペントフラノース(化合物18)の合成
【0184】
【化31】
【0185】
化合物17(1.10g、1.94mmol)をアセトニトリル(20.0mL)に溶解し、クロロチオノギ酸フェニル(0.40mL、2.89mmol)、4−ジメチルアミノピリジン(0.71g、5.81mmol)を加え、室温で3時間撹拌した。反応液を酢酸エチルで希釈後、有機層を0.1N塩酸、飽和重曹水で洗浄し、無水硫酸マグネシウム上で乾燥した。有機層を減圧下濃縮し、得られた粗チオ炭酸エステルを精製することなく次の反応に用いた。
粗チオ炭酸エステルをトルエンで3回共沸した後、トルエン(30.0mL)に溶解し、減圧脱気した。この溶液をアルゴン雰囲気下、80℃で水素化トリブチルスズ(2.61mL、9.70mmol)、少量のアゾビス(イソブチロニトリル)を加え、同条件で1時間撹拌した。反応液を減圧下濃縮後、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 100mL、ヘキサン:酢酸エチル=10:1)で精製し、化合物18(1.07g、1.94mmol、quant.)を得た。
【0186】
1H−NMR(CDCl3)δ:7.69−7.38(10H,m,aromatic),5.90(1H,d,H−1,J=4.00),4.85(1H,t,H−2,J=5.00),3.82(1H,d,H−5a,J=11.0),3.58(1H,d,H−5b,J=10.5),2.64(1H,dd,H−3a,J=6.00,14.0),2.40(1H,d,H−3b,J=14.0),1.68,1.36(each3H,s,acetonide),1.04(9H,s,t−Bu)0.92(9H,t,Et,J=8.00),0.54(6H,q,Et,J=8.00).
【0187】
(4)1,2−ジ−O−アセチル−5−O−t−ブチルジフェニルシリル−3−デオキシ−4−C−トリエチルシリルエチニル−D−キシロ−ペントフラノース(化合物19)の合成
【0188】
【化32】
【0189】
化合物18(1.07g、1.94mmol)を80%酢酸(100mL)に溶解し、トリフルオロ酢酸(10.0mL)を加え、40℃で3時間撹拌した。反応液を減圧下濃縮後、残さをトルエンで共沸し、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 100mL、ヘキサン−酢酸エチル=4:1)で精製した。得られた残さをピリジン(20.0mL)に溶解し、無水酢酸(0.49mL)を加え、室温で終夜撹拌した。反応液を減圧下濃縮後、残さをトルエンで共沸し、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 100mL、ヘキサン:酢酸エチル=5:1)で精製した。化合物19(0.75g、)を得た。
【0190】
1H−NMR(CDCl3)δ:7.71−7.37(10H,m,aromatic),6.44(0.3H,d,H−1−alpha,J=4.50),6.30(0.7H,s,H−1−beta),5.36(0.3H,m,H−2−alpha),5.19(0.7H,d,H−2−beta,J=5.50),3.77,3.74(each0.7H,d,H−5−beta,J=10.0),3.76,3.62(each0.3H,d,H−5−alpha,J=11.0),2.86(0.3H,dd,H−3a−alpha,J=8.50,12.5),2.72(0.7H,dd,H−3a−beta,J=5.50,14.0),2.39(0.3H,dd,H−3b−alpha,J=10.0,12.5),2.33(0.7H,d,H−3b−beta,J=14.0),2.11,2.08(each0.9H,s,acety−alpha),2.10,1.80(each2.1H,s,acetyl−beta),1.074(6.3H,s,t−Bu−beta),1.067(2.7H,s,t−Bu−alpha),0.97(6.3H,t,Et−beta,J=8.00),0.94(2.7H,t,Et−alpha,J=8.00),0.58(4.2H,q,Et−beta,J=8.00),0.54(1.8H,q,Et−alpha,J=8.00).
【0191】
(5)9−(2−O−アセチル−5−O−t−ブチルジフェニルシリル−3−デオキシ−4−C−トリエチルシリルエチニル−β−D−キシロ−ペントフラノシル)−2,6−ジアミノプリン(化合物20)の合成
【0192】
【化33】
【0193】
2,6−ジアミノプリン(1.21g、8.06mmol)をアセトニトリル(24.0mL)に懸濁し、N,O−ビス(トリメチルシリル)アセトアミド(11.9mL、48.1mmol)を加え、80℃で3時間撹拌した。この溶液を減圧下濃縮後、残さを1,2−ジクロロエタンで3回共沸した。残さに化合物19(2.39g、4.02mmol)の1,2−ジクロロエタン溶液(24.0mL)、トリフルオロメタンスルホン酸トリメチルシリル(3.05mL、16.9mmol)を加え、アルゴン雰囲気下、50℃で5時間、80℃で10時間撹拌した。飽和重曹水を加え撹拌後、溶液をセライトろ過した。有機層を無水硫酸マグネシウム上で乾燥後、減圧下濃縮し、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 300mL、クロロホルム:メタノール=20:1)で精製した。ヘキサン−酢酸エチルから結晶化し、化合物20(1.60g、2.34mmol、58.2%)を得た。
【0194】
1H−NMR(CDCl3)δ:7.67−7.33(10H,m,aromatic),6.13(1H,d,H−1’,J=3.50),5.77(1H,md,H−2’),5.28(2H,bs,NH2),4.49(2H,bs,NH2),3.67(1H,d,H−5’a,J=10.5),3.79(1H,d,H−5’b,J=11.0),3.18(1H,dd,H−3’a,J=7.50,14.0),2.37(1H,dd,H−3’b,J=3.00,14.0),1.06(9H,s,t−Bu),0.99(9H,t,Et,J=8.00),0.61(6H,q,Et,J=8.00).
【0195】
(6)5’−O−t−ブチルジメチルシリル−3’−デオキシ−4’−C−トリエチルシリルエチニル−2−フルオロアデノシン(化合物21)の合成
【0196】
【化34】
【0197】
化合物20(100mg、0.146mmol)をピリジン(3.00mL)に溶解し、−15℃でフッ化水素−ピリジン(7.00mL)、亜硝酸t−ブチル(160μl、1.34mmol)を加え、同温度で30分間撹拌した。反応液に水を加えた後、酢酸エチルで抽出した。有機層を飽和重曹水で洗浄後、無水硫酸マグネシウム上で乾燥し、減圧下濃縮した。残さをシリカゲルカラムクロマトグラフィー(シリカゲル 300mL、クロロホルム:メタノール=100:1〜20:1)で精製した。得られた残さ(48.9mg)をジクロロメタン(1.30mL)に溶解し、0℃でトリフルオロメタンスルホン酸t−ブチルジメチルシリル(36.0μl、0.157mmol)、コリジン(44.1μl、0.0331mmol)を加え、同温度で50分間撹拌した。反応液に水を加え、クロロホルムで抽出した。有機層を0.01N塩酸、飽和重曹水で洗浄後、無水硫酸マグネシウム上で乾燥し、減圧下濃縮した。残さをジオキサン(3.00mL)に溶解し、28%アンモニア水(0.30mL)を加え、室温で30分間撹拌した。反応液を減圧下濃縮後、残さをメタノール(1.20mL)に溶解し、28%アンモニア水(0.80mL)を加え、室温で2時間撹拌した。反応液を減圧下濃縮し、析出した結晶をろ取し、化合物21(34.0mg、0.0652mmol、44.7%)を得た。
【0198】
1H−NMR(CDCl3)δ:8.04(1H,s,H−8),5.99(1H,d,H−1’,J=4.00),5.80(2H,bs,NH2),4.73(1H,m,H−2’),4.23(1H,d,2’−OH,J=5.00),3.88(1H,d,H−5’a,J=11.0),3.70(1H,d,H−5’b,J=11.0),2.82(1H,dd,H−3’a,J=7.50,13.0),2.42(1H,dd,H−3’b,J=6.50,13.0),1.01(9H,t,Et,J=8.00),0.80(9H,s,t−Bu),0.64(6H,q,Et,J=8.00),0.039,−0.013(each3H,s,Me).
【0199】
(7)5’−O−t−ブチルジメチルシリル−2’,3’−ジデオキシ−4’−C−トリエチルシリルエチニル−2−フルオロアデノシン(化合物22)の合成
【0200】
【化35】
【0201】
化合物21(32.0mg、0.061mmol)をアセトニトリルで3回共沸後、アセトニトリル(1.00mL)に溶解し、クロロチオノギ酸フェニル(12.7μl、0.092mmol)、4−ジメチルアミノピリジン(22.5mg、0.180mmol)を加え、室温で1時間撹拌した。反応液を酢酸エチルで希釈後、有機層を0.01N塩酸、飽和重曹水で洗浄し、無水硫酸マグネシウム上で乾燥した。有機層を減圧下濃縮し、得られた粗チオ炭酸エステルを精製することなく次の反応に用いた。
粗チオ炭酸エステルをトルエンで3回共沸した後、トルエン(1.00mL)に溶解し、減圧脱気した。この溶液をアルゴン雰囲気下、80℃でトリス(トリメチルシリル)シラン(94.6μl、0.306mmol)、少量のアゾビス(イソブチロニトリル)を加え、同条件で1時間撹拌した。反応液を減圧下濃縮後、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 10mL、クロロホルム:メタノール=200:1〜100:1)で精製し、化合物22(26.1mg、0.0516mmol、84.6%)を得た。
【0202】
1H−NMR(CDCl3)δ:8.24(1H,s,H−8),6.36(1H,dd,H−1’,J=2.50,7.00),5.91(2H,bs,NH2),4.04(1H,d,H−5’a,J=11.0),3.81(1H,d,H−5’b,J=11.0
),2.83(1H,m,H−2’a),2.54(1H,m,H−3’a),2.37(1H,m,H−2’b),2.11(1H,m,H−3’b),1.00(9H,t,Et,J=8.00),0.93(9H,s,t−Bu),0.62(6H,q,Et,J=8.00),0.13(6H,s,Me).
【0203】
(8)2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン(化合物23)の合成
【0204】
【化36】
【0205】
化合物22(101mg、0.200mmol)をテトラヒドロフラン(10mL)に溶解し、1M フッ化テトラブチルアンモニウム−テトラヒドロフラン溶液(0.42mL、0.42mmol)を加え、室温で5分間撹拌した。反応液に酢酸(24μl)を加えた後、減圧下濃縮し、残さをシリカゲルカラムクロマトグラフィー(シリカゲル 15mL、クロロホルム:メタノール=40:1〜20:1)で精製した。化合物23(53.0mg、0.191mmol、95.7%)を得た。
【0206】
1H−NMR(MeOD)δ:8.23(1H,s,H−8),6.22(1H,dd,H−1’,J=4.00,7.00),3.77(1H,d,H−5’a,J=12.5),3.61(1H,d,H−5’b,J=12.0),2.94(1H,s,ethynyl),2.66(1H,m,H−2’a),2.54(1H,m,H−3’a),2.42(1H,m,H−2’b),2.11(1H,m,H−3’b).
【0207】
合成例8:2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン5’−H−ホスホネート(化合物24)の合成
【0208】
【化37】
【0209】
化合物23(20.0mg、0.07mmol)をピリジン(1mL)に溶解し、ホスホン酸(11.8mg,0.144mmol)、ジシクロヘキシルカルボジイミド(44.7mg、0.216mmol)を加え、室温で5時間撹拌した。反応液を減圧下濃縮し、残さをODSカラムクロマトグラフィー(ODS 10mL、0−1%アセトニトリル)により精製した。得られた残さをDowex 50Wx8カラム(Na型)に通過させた。通過液を濃縮してえられた残さをメタノール−エーテルで粉末とし、化合物24(4.7mg、13μmol、18.6%)を得た。
【0210】
1H−NMR(MeOD)δ:8.37(1H,s,H−8),6.77(1H,d,H−P,J=625),6.32(1H,dd,H−1’,J=4.00,6.50),4.09(1H,m,H−5’),2.71(2H,m,H−2’a,H−3’a,),2.52(1H,m,H−2’b),2.29(1H,m,H−3’b).
【0211】
31P−NMR(MeOD)δ:4.52
【0212】
製剤例1:錠剤
本発明化合物 30.0mg
微粉末セルロース 25.0mg
乳糖 39.5mg
スターチ 40.0mg
タルク 5.0mg
ステアリン酸マグネシウム 0.5mg
上記組成から常法によって錠剤を調製する。
【0213】
製剤例2:カプセル剤
本発明化合物 30.0mg
乳糖 40.0mg
スターチ 15.0mg
タルク 5.0mg
上記組成から常法によってカプセル剤を調製する。
【0214】
製剤例3:注射剤
本発明化合物 30.0mg
グルコース 100.0mg
上記組成を注射用精製水に溶解して注射剤を調製する。
【0215】
以下に試験例を示す。試験においては薬剤として以下の本発明化合物と公知化合物を使用した。
本発明化合物:
化合物4: 2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン
化合物8: 4’−C−シアノ−2’−デオキシ−2−フルオロアデノシン
化合物9: 2−クロロ−2’−デオキシ−4’−C−エチニル−アデノシン
化合物10:2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン5’−H−ホスホネート
化合物13:2’,3’−ジデヒドロ−2’,3’−ジデオキシ−4’−C−エチニル−2−フルオロアデノシン
公知化合物:
AZT:アジドチミジン
EdAdo:2’−デオキシ−4’−C−エチニルアデノシン
EdDAP:9−(4−C−エチニル−2−デオキシ−リボペントフラノシル)−2,6−ジアミノプリン
ddAdo:2’,3’−ジデオキシアデノシン
【0216】
試験例1
<方法>抗ヒト免疫不全ウイルス(HIV)活性
1)MT−4細胞を用いたMTT法
1.96ウエルプレートを用いて被験薬剤を希釈する(100μl)。ここに1ウエルあたり10000個になるようにHIV−1(IIIbstrain;100TCID50)感染及び非感染MT−4細胞を加えて、37℃で5日間培養する。
2.MTT(20μl、7.5mg/mL)を加えて更に2〜3時間培養する。
3.培養終了後、120μlをとり、MTT停止液(Triton X−100 4%、HCl 0.04Nを加えたイソプロパノール(Isopropanol))を加え、撹拌して生成したホルマザン(formazam)を溶解し、540nmにおける吸光度を測定する。この測定値は生細胞数に比例するため、感染MT−4細胞を用いた試験の測定値が50%になった被験薬剤の濃度をEC50、また非感染MT−4細胞を用いた試験の測定値が50%になった被験薬剤の濃度をCC50とする。
【0217】
2)HeLa CD4/LTR−beta−Gal細胞を用いたMAGIアッセイ
1.HeLa CD4/LTR−beta−Gal細胞を1ウエルあたり10000個の割合で96ウエルに加える。12〜24時間後に培養液を捨て、希釈した被験薬剤(100μl)を加える。
2.各種HIV株(野生株:WT、耐性株:MDR、M184V;各50TCID50相当)を加えて48時間培養する。
3.培養終了後、1%ホルムアルデヒド(formaldehyde)、0.2%グルタールアルデヒド(glutaraldehyde)を加えたPBSで細胞を5分間固定する。
4.PBSで3回洗浄後、0.4mg/mL X−Galで1時間染色し、青く染色された細胞の数を透過型実体顕微鏡下で各ウエルのプラーク数を数え、青染された細胞を50%減少させる被験薬剤の濃度をEC50とする。
【0218】
<結果>抗ヒト免疫不全ウイルス(HIV)活性及び細胞毒性
1)MT−4細胞を用いたMTT法
【0219】
【表1】
【0220】
2)HeLa CD4/LTR−beta−Gal細胞を用いたMAGIアッセイ
【0221】
【表2】
【0222】
試験例2
<方法>化合物4のアデノシンデアミナーゼに対する安定性
0.5mMの化合物4(50mM Tris−HCl緩衝液溶液(pH7.5))0.5mLに子ウシ腸管由来アデノシンデアミナーゼ 0.01unitを加え、25℃でインキュベートする。
15分ごとに反応液5μlを取り、高速液体クロマトグラフィー(HPLC)により分析する。反応時間0分のときの被験薬剤のピーク面積を100%として、その経時変化を観察する。なお、HPLC分析は、以下の条件で行った。
カラム:YMC−Pack ODS−A(250×6.0mm)
溶出液:15% MeCN−50mM TEAA
流速: 1mL/min
温度: 30℃
検出: 260nm
【0223】
<結果>
その結果、図1に示すように、従来の2’−デオキシ−4’−C−エチニルアデノシン(EdAdo)は脱アミノされるに対し、本発明化合物の2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン(化合物4)は全く脱アミノされず、アデノシンデアミナーゼに対し、抵抗性を有することが明らかとなった。
【0224】
試験例3
<方法>化合物4の酸性条件下における安定性
化合物4(2.9mg)もしくは2’,3’−ジデオキシアデノシン(ddAdo:2.4mg)を予め37℃に暖めた試験液(塩化ナトリウム2.0gに塩酸7.0mL及び水を加えて溶かし1000mLとしたもの)10mLに溶解し、同温度でインキュベートする。
反応液100μlを取り、0.1規定水酸化ナトリウム水溶液で中和後、5μlをHPLCにより分析する。なお、HPLC分析条件は、試験例2と同じである。
【0225】
<結果>
その結果、従来のddAdoは本条件下5分ほどで約98%が分解されるのに対し(図3)、本発明化合物の2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン(化合物4)の分解は非常に遅く、酸性条件下比較的安定であることが明らかとなった(図2)。
【0226】
試験例4
<方法>化合物4のin vivoにおける急性毒性試験
6週齢、雄性、ICRマウスに被検薬剤(化合物4:生理食塩水に溶解又は懸濁)を一群8匹ずつ、100mg/kgまで経口あるいは静脈内投与し、7日間マウスの生死、体重を測定した。
【0227】
<結果>
化合物4は、単回投与では経口、静脈内いずれの投与経路でも100mg/kgまでマウスの死亡は認められなかった(下記表3)。また、図4に示すように体重減少は見られず、下痢のような症状も観察されなかったことから、本発明化合物の2’−デオキシ−4’−C−エチニル−2−フルオロアデノシン(化合物4)はマウスにおいて急性毒性を示さないことが明らかになった。
【0228】
【表3】
【特許請求の範囲】
【請求項1】
式[IV]
【化1】
(式中、R1はエチニル基又はシアノ基を示す)
で表される化合物の3’位水酸基及び5’位水酸基を保護し、式[V]
【化2】
(式中、Pは保護基を示し、R1は前記と同じ)
で表される化合物を得、該化合物[V]の2位アミノ基をフッ素原子又は塩素原子へと変換して[VI]
【化3】
(式中、Xはフッ素原子又は塩素原子を示し、R1及びPは前記と同じ)
で表される化合物を得、次いで該化合物の[VI]の保護基を除去することを特徴とする式[I]
【化4】
(式中、R2は水素原子を示し、R1及びXは前記と同じ)
で表される化合物の製造法。
【請求項2】
Pで示される保護基が、エーテル系保護基、アシル系保護基、シリル系保護基又はアセタール系保護基である請求項1記載の製造法。
【請求項3】
化合物[V]の2位アミノ基をフッ素原子又は塩素原子に変換する手段が、亜硝酸誘導体で処理後、フッ素化試薬又は塩素化試薬を反応させる手段か、あるいは2,6位のアミノ基を亜硝酸誘導体で処理して2,6−ジフルオロプリン誘導体又は2,6−ジクロロプリン誘導体とした後、アンモニア処理により塩基部6位のフッ素原子又は塩素原子をアミノ基へとする手段である請求項1又は2記載の製造法。
【請求項4】
化合物[V]の2位のアミノ基をフッ素原子又は塩素原子に変換する手段が、亜硝酸誘導体で処理後、フッ素化試薬又は塩素化試薬を反応させる手段である請求項1又は2記載の製造法。
【請求項5】
化合物[V]の2位アミノ基をフッ素原子又は塩素原子に変換する手段が、テトラフルオロホウ酸中の亜硝酸ナトリウム、フッ化水素ピリジン中の亜硝酸エステル、有機溶媒中の三塩化アンチモン−亜硝酸t−ブチル、又は有機溶媒中の塩化アセチル−亜硝酸ベンジルトリエチルアンモニウムを反応させる手段である請求項4記載の製造法。
【請求項1】
式[IV]
【化1】
(式中、R1はエチニル基又はシアノ基を示す)
で表される化合物の3’位水酸基及び5’位水酸基を保護し、式[V]
【化2】
(式中、Pは保護基を示し、R1は前記と同じ)
で表される化合物を得、該化合物[V]の2位アミノ基をフッ素原子又は塩素原子へと変換して[VI]
【化3】
(式中、Xはフッ素原子又は塩素原子を示し、R1及びPは前記と同じ)
で表される化合物を得、次いで該化合物の[VI]の保護基を除去することを特徴とする式[I]
【化4】
(式中、R2は水素原子を示し、R1及びXは前記と同じ)
で表される化合物の製造法。
【請求項2】
Pで示される保護基が、エーテル系保護基、アシル系保護基、シリル系保護基又はアセタール系保護基である請求項1記載の製造法。
【請求項3】
化合物[V]の2位アミノ基をフッ素原子又は塩素原子に変換する手段が、亜硝酸誘導体で処理後、フッ素化試薬又は塩素化試薬を反応させる手段か、あるいは2,6位のアミノ基を亜硝酸誘導体で処理して2,6−ジフルオロプリン誘導体又は2,6−ジクロロプリン誘導体とした後、アンモニア処理により塩基部6位のフッ素原子又は塩素原子をアミノ基へとする手段である請求項1又は2記載の製造法。
【請求項4】
化合物[V]の2位のアミノ基をフッ素原子又は塩素原子に変換する手段が、亜硝酸誘導体で処理後、フッ素化試薬又は塩素化試薬を反応させる手段である請求項1又は2記載の製造法。
【請求項5】
化合物[V]の2位アミノ基をフッ素原子又は塩素原子に変換する手段が、テトラフルオロホウ酸中の亜硝酸ナトリウム、フッ化水素ピリジン中の亜硝酸エステル、有機溶媒中の三塩化アンチモン−亜硝酸t−ブチル、又は有機溶媒中の塩化アセチル−亜硝酸ベンジルトリエチルアンモニウムを反応させる手段である請求項4記載の製造法。
【図1】

【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公開番号】特開2011−256173(P2011−256173A)
【公開日】平成23年12月22日(2011.12.22)
【国際特許分類】
【出願番号】特願2011−142793(P2011−142793)
【出願日】平成23年6月28日(2011.6.28)
【分割の表示】特願2007−315315(P2007−315315)の分割
【原出願日】平成17年3月24日(2005.3.24)
【出願人】(000006770)ヤマサ醤油株式会社 (56)
【Fターム(参考)】
【公開日】平成23年12月22日(2011.12.22)
【国際特許分類】
【出願日】平成23年6月28日(2011.6.28)
【分割の表示】特願2007−315315(P2007−315315)の分割
【原出願日】平成17年3月24日(2005.3.24)
【出願人】(000006770)ヤマサ醤油株式会社 (56)
【Fターム(参考)】
[ Back to top ]