
NMRによる定量分析を用いた分配係数の測定方法

【課題】従来の分配係数の測定法は、水相中及び有機相中の化合物の定量のために検量線の作成が必要である。しかしながら、検量線の作成には多量の化合物が必要であるため、環境への負荷が大きい。また、作業負荷が大きいことから簡便性、迅速性に欠ける点も課題である。また、UV検出器を備えたHPLC又は紫外可視吸光光度計で測定する場合、紫外吸収が弱い若しくは全くない化合物には使用できない点も課題である。
【解決手段】本発明は、水相中及び有機相中の化合物の定量にNMRを用いることで上記課題を解決する。
【解決手段】本発明は、水相中及び有機相中の化合物の定量にNMRを用いることで上記課題を解決する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、NMRによる定量分析工程を含む、分配係数の測定方法に関する。
【背景技術】
【0002】
分配係数は分配比、分配率などともよばれ、化合物の疎水性を表す数値である。分配係数は化合物が水と有機溶媒の二相系に溶解したときの平衡溶解度比であり、下記式(1)
【0003】
【数1】

【0004】
[式中、Pは分配係数、Coは有機相中の化合物の濃度、Cwは水相中の化合物の濃度を表す]
で表される。一般的には、Pの常用対数である下記式(2)
【0005】
【数2】
【0006】
[式中、P、Co、Cwは前述と同義]
が分配係数として広く用いられている。分配係数は化合物の脂溶性を表すため、特に医薬品、農薬及び環境物質の体内吸収や残留性の指標となっている。例えば、化審法では、化合物の物性の指標として1−オクタノールと水の二相系での分配係数が採用されている。また、分配係数の測定法も日本工業規格(非特許文献1)やOECD Test Guideline(非特許文献2)によって標準化されている。
【0007】
ところで、今日用いられている分配係数の測定法(以下、従来法)では、有機相や水相中の化合物濃度を高速液体クロマトグラフィー(HPLC)や紫外可視吸光光度計で定量するため、検量線の作成が不可欠となる。検量線の作成には多量の化合物が必要であるが、化合物が希少である場合や環境への負荷を考慮した場合、化合物の使用量は少ないことが望ましい。また、検量線の作成は作業負荷が大きいため、種々の化合物の分配係数を測定する場合は特に問題となる。さらに従来法は、UVで化合物を定量する測定機器を用いる場合、HPLCや紫外可視吸光光度計で化合物を定量するため、紫外吸収が弱いか、もしくは全くない化合物には使用できないという欠点がある。
【0008】
また、化合物の分配係数を実測するのではなく、コンピューターを用いて分配係数を計算する方法も報告されているが、計算値は実測値と乖離することがあるため、信頼性の面から実測することが好ましい。
【0009】
ところで、近年、NMRを用いた化合物の定量分析法が報告されている(非特許文献3)。NMRを用いた定量分析法のひとつである内部標準法では、化合物と内標準物質の両方を含有する溶液をNMRで測定し、NMRスペクトル上の化合物と内標準物質のそれぞれのシグナル面積を比較することで、化合物の定量を可能としている。
【先行技術文献】
【非特許文献】
【0010】
【非特許文献1】日本工業規格Z7260−107(2000)、「分配係数(1−オクタノール/水)の測定−フラスコ振とう法」.
【非特許文献2】OECD Test Guideline、(OECD理事会決定「C(81)30最終別添1」)107.
【非特許文献3】平成21年度成果報告書 基準認証研究開発事業「1対多型校正技術の研究開発」 平成22年3月経済産業省産業技術環境局知的基盤課 委託先(独)産業技術総合研究所
【発明の概要】
【発明が解決しようとする課題】
【0011】
従来の分配係数の測定法は、水相中及び有機相中の化合物の定量のために検量線の作成が必要である。しかしながら、検量線の作成には多量の化合物が必要であるため、環境への負荷が大きい。また、作業負荷が大きいことから簡便性、迅速性に欠ける点も課題である。また、UV検出器を備えたHPLC又は紫外可視吸光光度計で測定する場合、紫外吸収が弱い若しくは全くない化合物には使用できない点も課題である。
【課題を解決するための手段】
【0012】
本発明は、水相中及び有機相中の化合物の定量にNMRを用いることで上記課題を解決した(以下、NMR法)。すなわち、本発明は以下の通りである。
【0013】
第1発明は、NMRを用いた定量分析工程を含む、分配係数の測定方法に関する。
第2発明は、以下の工程を含む分配係数の測定方法であって、
i)化合物、重水及び重水素化有機溶媒を混合する工程、
ii)工程iの混合物を水相及び有機相に分離する工程、及び
iii)工程iiの水相及び/又は有機相中の化合物の特定基のシグナル面積をNMRを用いて測定する工程、
を含む、第1発明記載の分配係数の測定方法に関する。
第3発明は、工程iiiが、水相及び有機相中の化合物の特定基のシグナル面積をNMRを用いて測定する工程である、第2発明記載の分配係数の測定方法に関する。
【0014】
第4発明は、以下の工程を含む分配係数の測定方法であって、
i)化合物、飽和重水及び飽和重水素化有機溶媒を混合する工程、
ii)工程iの混合物を水相及び有機相に分離する工程、
iii)工程iiで得た有機相に内標準物質を溶解させ有機相試料溶液とした後、NMRを用いて有機相試料溶液中の化合物及び内標準物質の特定基のシグナル面積を測定し、当該シグナル面積を用いて有機相中の化合物含量を算出する工程、
iv)工程iiで得た水相に工程iiiと同一又は異なる内標準物質を溶解させ水相試料溶液とした後、NMRを用いて水相試料溶液中の化合物及び内標準物質の特定基のシグナル面積を測定し、当該シグナル面積を用いて水相中の化合物含量を算出する工程、及び
v)工程iiiで算出した有機相中の化合物含量と、工程ivで算出した水相中の化合物含量を用いて化合物の分配係数を算出する工程、
を含む、第3発明記載の分配係数の測定方法に関する。
【0015】
第5発明は、以下の工程を含む分配係数の測定方法であって、
i)化合物、飽和重水及び飽和重水素化有機溶媒を混合する工程、
ii)工程iの混合物を水相及び有機相に分離する工程、
iii)工程iiで得た水相に重水素化酸、重水素化塩基若しくはシフト試薬を溶解させ水相試料溶液とし、工程iiで得た有機相を有機相試料溶液とするか、或いは工程iiで得た水相を水相試料溶液とし、工程iiで得た有機相にシフト試薬を溶解させ有機相試料溶液とし、当該有機相試料溶液と水相試料溶液のうち一方を二重NMR管の外管に採取し、他方を二重NMR管の内管に採取した後、NMRを用いて有機相試料溶液及び水相試料溶液中の化合物の特定基のシグナル面積を測定する工程、及び
iv)工程iiiで測定した有機相試料溶液及び水相試料溶液中の化合物の特定基のシグナル面積を用いて化合物の分配係数を算出する工程、
を含む、第3発明記載の分配係数の測定方法に関する。
【発明の効果】
【0016】
本発明によれば、検量線の作成が不要であること等から、従来法と比較して少量の化合物で分配係数を測定することが可能である。また、簡便、迅速に分配係数を測定することが可能である。さらに、本発明によれば、紫外吸収を示さない化合物の分配係数の測定も可能である。
【図面の簡単な説明】
【0017】
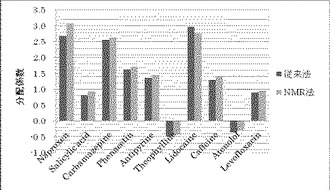
【図1】横軸に化合物の種類、縦軸に化合物の分配係数をとり、従来法とNMR法で測定した各種化合物の分配係数を示したグラフである。
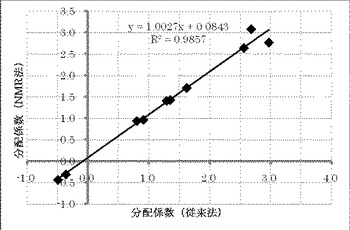
【図2】横軸に分配係数(従来法)、縦軸に分配係数(NMR法)をとり、従来法とNMR法で測定した分配係数の相関性を示したグラフである。
【発明を実施するための形態】
【0018】
本明細書中において使用される語句の定義は以下の通りである。
【0019】
化合物とは、酸性化合物、中性化合物、塩基性化合物、両性化合物等の任意の化合物を表す。なお、本発明においては、コンピューター(Pallas3.0、インフォコム株式会社)を用いて計算し、酸塩基解離定数が6以下の化合物を酸性化合物、7.8以上の化合物を塩基性化合物、両方の性質を持ち合わせるものを両性化合物とし、それ以外を中性化合物と分類した。
【0020】
重水とは、水分子の二つの水素原子を重水素原子に置き換えた水を表し、D2Oと表すこともある。
【0021】
重水素化有機溶媒とは、有機溶媒分子に含まれる水素原子の一部または全部を重水素原子に置き換えた有機溶媒を表す。重水素化有機溶媒としては、例えば、シクロヘキサン−d12、トルエン−d8、ベンゼン−d6、クロロホルム−d、ジクロロメタン−d2又は1−オクタノール−d18が挙げられる。
【0022】
内標準物質とは、化合物を定量するために、水相及び/又は有機相に添加する物質である。内標準物質としては、例えば、2,6−ジクロロベンズアルデヒド、2−ヒドロキシ−3,5−ジニトロ−安息香酸、テレフタル酸ジメチルエステル、フタル酸水素カリウム、安息香酸、1,3,5−トリオキサン、テトラメチルピラジン、ジメチルスルホン、酢酸ナトリウム、トリメチルシリルプロパンスルホン酸ナトリウム−d6、トリメチルシリルプロピオン酸ナトリウム−d4又は1,4−ビス(トリメチルシリル)ベンゼン−d4が挙げられる。
【0023】
シグナル面積は、ピーク面積又は積分値と同義である。
【0024】
特定基とは、化合物および内標準物質のそれぞれの任意の置換基を表す。
【0025】
飽和重水素化有機溶媒とは、重水が飽和した重水素化有機溶媒を表す。
【0026】
飽和重水とは、重水素化有機溶媒が飽和した重水を表す。
【0027】
重水素化酸とは、酸分子に含まれる水素原子の一部または全部を重水素原子に置き換えた酸を表す。重水素化酸としては、例えば、蟻酸−d2、酢酸−d、酢酸−d4、トリフルオロ酢酸−d、塩酸−d(重塩酸)、硫酸−d2が挙げられる。
【0028】
重水素化塩基とは、塩基分子に含まれる水素原子の一部または全部を重水素原子に置き換えた塩基を表す。重水素化塩基としては、例えば、水酸化アンモニウム−d5、ピリジン−d5、水酸化ナトリウム−d、水酸化カリウム−dが挙げられる。
【0029】
シフト試薬とは、有機配位子に配位結合した希土(ランタニド)類系列のイオンである。例えば、トリス(2、2、6、6−テトラメチル−3、5−ヘプタンジオナト)ユウロピウム (Eu(dpm)3)、トリス(6、6、7、7、8、8、8−ヘプタフルオロ−2、2−ジメチル−3、5−オクタンジオナト)ユウロピウム (Eu(fod)3)、トリス[N、N−ビス(トリメチルシリル)アミド]ユウロピウム、トリス(4、4、4−トリフルオロ−1−(2−チエニル)−1、3−ブタンジオノ)ユウロピウム、トリス(6、6、7、7、8、8、8−ヘプタフルオロ−2、2−ジメチル−3、5−オクタンジオナト)イッテルビウム (Yb(fod)3)、トリス(2、2、6、6−テトラメチル−3、5−ヘプタンジオナト)ジスプロシウム(III) (Dy(dpm)3)、トリス(2、2、6、6‐テトラメチル‐3、5‐ヘプタンジオナト)ガドリニウム (Gd(dpm)3)が挙げられる。
【0030】
二重NMR試料管とは、外管(アウターチューブ)と内管(インナーチューブ)を備えたNMR管を表す。外管と内管の各々に試料溶液を採取することができる。
【0031】
本発明の好ましい形態は以下の通りである。
【0032】
化合物は、中性化合物及び両性化合物が、濃度による分配係数への影響が少ないため好ましい。また、より正確な分配係数が得られるため、LogPが−3〜3の範囲の化合物が好ましく、−2.5〜2.5の範囲の化合物が好ましく、−2〜2の範囲の化合物が特に好ましい。
【0033】
化合物の濃度は、重水素化有機溶媒1mLに対し、好ましくは0.008〜30 mgであり、更に好ましくは0.02〜10 mgであり、特に好ましくは1.8〜10 mgである。また、重水1mLに対し、好ましくは0.008〜30mgであり、更に好ましくは0.02〜10mgであり、特に好ましくは1.1〜10mgである。
【0034】
NMRは、測定感度の点からプロトンNMR(1H−NMR)が好ましい。
【0035】
重水素化有機溶媒は、有機溶媒分子に含まれる水素原子の全部を重水素原子に置き換えた有機溶媒が好ましい。好ましくはシクロヘキサン−d12、トルエン−d8、ベンゼン−d6、クロロホルム−d、ジクロロメタン−d2又は1−オクタノール−d18であり、更に好ましくはクロロホルム−d又は1−オクタノール−d18であり、特に好ましくはクロロホルム−dである。
【0036】
内標準物質は、そのNMRシグナルのうち少なくとも1つのシグナルが、化合物のNMRシグナルと重ならないものであれば、任意に選ぶことができる。有機相に添加する場合、好ましくは2,6−ジクロロベンズアルデヒド又は1,4−ビス(トリメチルシリル)ベンゼン−d4である。水相に添加する場合、好ましくはトリメチルシリルプロピオン酸ナトリウム-d4又はトリメチルシリルプロパンスルホン酸ナトリウム−d6である。
【0037】
重水素化酸は、好ましくは蟻酸−d2、酢酸−d、酢酸−d4、トリフルオロ酢酸−d、塩酸−d(重塩酸)、硫酸−d2であり、更に好ましくは酢酸−d4、トリフルオロ酢酸−d、塩酸−d(重塩酸)であり、特に好ましくは塩酸−d(重塩酸)である。
【0038】
重水素化塩基は、好ましくは水酸化アンモニウム−d5、ピリジン−d5、水酸化ナトリウム−d、水酸化カリウム−dである。
【0039】
シフト試薬は、好ましくはトリス(2、2、6、6−テトラメチル−3、5−ヘプタンジオナト)ユウロピウム、トリス(6、6、7、7、8、8、8−ヘプタフルオロ−2、2−ジメチル−3、5−オクタンジオナト)ユウロピウムである。
【0040】
特定基は任意に選択できるが、精確なシグナル面積を得るため、そのNMRシグナルが他のNMRシグナルと完全に分離している置換基から選ぶことが好ましい。
【0041】
以下に本発明の一般的な実施形態を説明するが、本実施形態によって本発明の範囲が限定されるものではない。
【0042】
(工程1)飽和溶媒の作成
適当量の重水素化有機溶媒と重水を激しく振り混ぜた後、室温下、24時間以上静置する。その後、二つの層を分離し、飽和重水素化有機溶媒と飽和重水を得る。
【0043】
(工程2)化合物の分配操作
化合物を精秤し、これに工程1で得られた飽和重水素化有機溶媒を一定量正確に加えて化合物を完全に溶解させる。これに工程1で得られた飽和重水を、飽和重水素化有機溶媒と同容量加え、激しく混合後、25℃で24時間振とうする。これを遠心分離により二つの層に分離し、有機相と水相を得る。
【0044】
(工程3)NMRによる、有機相及び/又は水相中の化合物の定量分析と分配係数の算出
有機相及び水相中の化合物の定量分析は、以下に示す個別定量法又は同時定量法により行うことができる。
【0045】
1.個別定量法
本方法では、工程2で得られた有機相及び/又は水相に内標準物質を添加し、得られた有機相試料溶液及び水相試料溶液についてNMRを用いて個別に定量分析を行った後、分配係数を算出する。
【0046】
1)有機相中の化合物の定量分析
内標準物質を精秤し、これに工程2で得られた有機相を一定量正確に加えて内標準物質を完全に溶解させ、これを有機相試料溶液とする。その後、有機相試料溶液をNMRで測定する。この際、公知技術(平成21年度成果報告書 基準認証研究開発事業「1対多型校正技術の研究開発」 平成22年3月経済産業省産業技術環境局知的基盤課 委託先(独)産業技術総合研究所)等を参考にして測定条件を選択し、化合物及び内標準物質の特定基のシグナル面積を得る。下記式(3)
【0047】
【数3】
【0048】
に化合物及び内標準物質の特定基のシグナル面積、特定基のプロトン数、分子量及びNMR測定に用いた質量並びに内標準物質の純度を代入することにより、有機相中の化合物の含量を算出する。
【0049】
2)水相中の化合物の定量分析
工程2で得られた水相中の化合物の定量分析は、以下に示す実測法又は差し引き法により行うことができる。適用可能な分配係数の範囲が広いこと及び正確な分配係数を得られることから実測法が好ましい。
<実測法>
工程2で得られた水相と、内標準物質を用いて、1)と同様の方法により水相試料溶液を作成後、NMRで測定し、式(3)に従って水相中の化合物含量を算出する。
<差し引き法>
下記式(4)
【0050】
【数4】
【0051】
により水相中の化合物含量を算出することができる。ここで、分配前の化合物含量とは、工程2の分配前の化合物純度を表す。例えば、工程2と同量の化合物と、一定量の内標準物質を精量し、これに工程2と同量の飽和重水素化有機溶媒を加えて完全に溶解させ試料溶液とした後、1)と同様の方法によりNMRを測定し、式(3)に従って算出する。このようにして得た分配前の化合物含量と、1)で得た有機相中の化合物含量を式(4)に代入し、水相中の化合物含量を算出する。
【0052】
3)分配係数の算出
化合物含量の比は化合物濃度の比と等しいため、式(2)のCoに1)で算出した有機相中の化合物含量を代入し、Cwに2)で算出した水相中の化合物含量を代入することで、化合物の分配係数を算出する。
【0053】
2.同時定量法
本方法では、工程2で得られた有機相又は水相に、重水素化酸、重水素化塩基若しくはシフト試薬を加え、得られた有機相試料溶液及び水相試料溶液の一方を二重NMR試料管の外管に、他方を二重NMR試料管の内管に採取し、NMRを用いて定量分析を行った後、分配係数を算出する。本測定法は内標準物質を必要とせず、また、有機相試料溶液と水相試料溶液を同時に測定できるため、簡便である。
【0054】
1)補正値の算出
同一の化合物濃度を有する有機相試料溶液と水相試料溶液であっても、一方を二重NMR管の外管に、他方を二重NMR管の内管に採取し、NMR測定した場合、種々の要因により化合物の特定基のシグナル面積に差異が生じる。したがって、当該差異を補正するための補正値を算出する。
化合物を重水素化有機溶媒に溶解し有機相試料溶液とする。また、有機相試料溶液中の化合物濃度と等しくなるように化合物及び重水素化酸、重水素化塩基若しくはシフト試薬を重水に溶解し水相試料溶液とする。或いは化合物及びシフト試薬を重水素化有機溶媒に溶解し有機相試料溶液とする。また、有機相試料溶液中の化合物濃度と等しくなるように化合物を重水に溶解し水相試料溶液とする。
有機相試料溶液を二重NMR試料管の外管に、水相試料溶液を二重NMR試料管の内管に採取し、NMRを測定し、水相試料溶液及び有機相試料溶液中の化合物の特定基のシグナル面積を得る。溶媒量、化合物の質量及びシグナル面積を下記式(5)
【0055】
【数5】
【0056】
に代入し、補正値を算出する。
【0057】
2)化合物の定量分析
工程2で得られた水相に重水素化酸、重水素化塩基若しくはシフト試薬を加えて水相試料溶液とし、工程2で得られた有機相はそのまま有機相試料溶液とする。或いは工程2で得られた水相はそのまま水相試料溶液とし、工程2で得られた有機相にシフト試薬を加えて有機相試料溶液とする。有機相試料溶液を二重NMR試料管の外管に、水相試料溶液を二重NMR試料管の内管に入れ、NMRを測定し、水相試料溶液及び有機相試料溶液中の化合物の特定基のシグナル面積を得る。
【0058】
3)分配係数の算出
化合物の特定基のシグナル面積は化合物濃度に比例する。したがって、シグナル面積の比は化合物濃度の比と等しい。すなわち、分配係数は下記式(6)
【0059】
【数6】
【0060】
により算出できる。
ここで、2)において水相に加える重水素化酸、重水素化塩基若しくはシフト試薬が水溶液の場合、水相は当該水溶液により希釈されていることから、水相試料溶液中の化合物の特定基のシグナル面積は、水相中の化合物の特定基のシグナル面積よりも希釈率分だけ小さい値となる。また、1)で述べたように、有機相試料溶液中の化合物の特定基のシグナル面積は補正する必要がある。これらを考慮し、分配係数は下記式(7)
【0061】
【数7】
【0062】
[式中、P、Io’、Iw’は前述と同義]
により算出できる。式(7)に1)で得られた補正値、2)で得られた化合物の特定基のシグナル面積、及び希釈率を代入することで、化合物の分配係数を算出する。なお、水相に加える重水素化酸、重水素化塩基若しくはシフト試薬が水溶液ではない場合は、希釈率は考慮しなくてよい。
【0063】
以下に本発明を実施例及び比較例にて更に詳細に説明するが、これらによって本発明の範囲が限定されるものではない。
【実施例】
【0064】
<比較例1>
従来法による、アンチピリンの分配係数測定
【0065】
1.検量線の作成
以下、クロロホルム溶液及び水溶液中のアンチピリン濃度に関する検量線を作成した。
1)クロロホルム溶液中のアンチピリン濃度に関する検量線
アンチピリン(シグマ・アルドリッチ製、10.67mg)にクロロホルム(関東化学製、特級)を加えて25mLとし、原液(426.8μg/mL)とした。原液2mLにクロロホルムを加えて20mLとし、溶液Aとした(42.68μg/mL)。溶液A4mLにクロロホルムを加えて20mLとし、溶液Bとした(8.536μg/mL)。溶液B4mLにクロロホルムを加えて20mLとし、溶液Cとした(1.707μg/mL)。溶液A7mLにクロロホルムを加えて10mLとし、溶液Dとした(29.88μg/mL)。溶液B4mLにクロロホルムを加えて10mLとし、溶液Eとした(3.414μg/mL)。溶液C4mLにクロロホルムを加えて10mLとし、溶液Fとした(0.6829μg/mL)。
溶液AからFを紫外可視吸光光度計で測定し、約278nmにおける吸光度について、x軸を濃度、y軸を吸光度とした回帰式を作成し、相関係数1.000、切片0.001564、傾き0.05176の下記式(8)
【0066】
【数8】
【0067】
を得た。
【0068】
2)水溶液中のアンチピリン濃度に関する検量線
アンチピリン(シグマ・アルドリッチ製、20.44mg)に超純水(ミリQ水)を加えて50mLとし、原液(408.8μg/mL)とした。原液2mLに超純水を加えて20mLとし、溶液Aとした(40.88μg/mL)。溶液A4mLに超純水を加えて20mLとし、溶液Bとした(8.176μg/mL)。溶液B4mLに超純水を加えて20mLとし、溶液Cとした(1.635μg/mL)。溶液A5mLに超純水を加えて10mLとし、溶液Dとした(20.44μg/mL)。溶液B4mLに超純水を加えて10mLとし、溶液Eとした(3.270μg/mL)。溶液C4mLに超純水を加えて10mLとし、溶液Fとした(0.6541μg/mL)。
溶液AからFを紫外可視吸光光度計で測定し、約242nmにおける吸光度について、x軸を濃度、y軸を吸光度とした回帰式を作成し、相関係数1.000、切片0.001521、傾き0.04860の下記式(9)
【0069】
【数9】
【0070】
を得た。
【0071】
2.分配係数測定
以下、クロロホルム/水系での分配係数を従来法により測定した。
超純水(ミリQ水、200mL)とクロロホルム(関東化学製、特級、200mL)を分液漏斗で振り混ぜた後、24時間以上放置し、上層と下層を分離した。得られた上層を飽和水、下層を飽和クロロホルムとした。
アンチピリン(シグマ・アルドリッチ製、201.27mg)に飽和クロロホルムを加えて20mLとし,アンチピリン溶液とした。アンチピリン溶液12mLに飽和水12mLを加えて室温で30分間激しく振とう後、25℃で24時間振とうし、遠心分離(3000rpm、10分)して上層(水相)と下層(有機相)を完全に分離した。得られた上層(水相)4mLに超純水を加えて20mLとし、溶液Aとした。溶液A 4mLに超純水を加えて20mLとし、水相試料溶液とした。また、得られた下層(有機相)2mLにクロロホルムを加えて10mLとし、溶液Bとした。溶液B 2mLにクロロホルムを加えて20mLとし、溶液Cとした。溶液C 2mLにクロロホルムを加えて20mLとし、有機相試料溶液とした。
有機相試料溶液と水相試料溶液の吸光度を紫外可視吸光光度計で測定した(有機相試料溶液:278nm、水相試料溶液:242nm)。それぞれの試料溶液から得られた吸光度を式(8)及び式(9)に代入し、有機相試料溶液中のアンチピリン濃度(Co)と水相試料溶液中のアンチピリン濃度(Cw)を定量した。得られたCo及びCwを式(2)に代入し、LogP=1.35を得た。
【0072】
<比較例2〜10>
その他9化合物についても比較例1と同様にして分配係数を測定した。
【0073】
<実施例1>
NMR法(実測法)による、アンチピリンの分配係数測定
重水(アクロス製、17mL)とクロロホルム−d(アクロス製、0.03v/v%TMS含有、17mL)を分液漏斗で振り混ぜた後、24時間以上放置し、上層と下層を分離した。得られた上層を飽和重水、下層を飽和クロロホルム−dとした。
アンチピリン(シグマ・アルドリッチ製、21.79mg)に飽和クロロホルム−d2mLを加えて溶解し、さらに飽和重水2mLを加えて30秒間激しく混合後、25℃で24時間振とうし、遠心分離(3000rpm、10分)して上層(水相)と下層(有機相)を完全に分離した。
得られた下層(有機相)1mLに内標準物質として2,6−ジクロロベンズアルデヒド(アクロス製、10.84mg)を溶解し、有機相試料溶液とした。得られた有機相試料溶液について表1の条件にて1H−NMRを測定した結果、アンチピリンの特定基のシグナル面積は3.00、2,6−ジクロロベンズアルデヒドの特定基のシグナル面積は1.10であった。なお、アンチピリンの特定基は1,2−ジヒドロ−3H−ピラゾール−3−オンの5位のメチル基、2,6−ジクロロベンズアルデヒドの特定基はホルミル基を選択した。
【0074】
表1 1H−NMR測定条件及び解析条件
【0075】
【表1】
【0076】
得られた結果に式(3)を適用して、有機相中のアンチピリン含量(Psample)を算出した。式(3)にIsample=3.00、Hsample=3、Msample=188.23、Wsample=10.90、Istd=1.10、Hstd=1、Mstd=175.01、Wstd=10.84、Pstd=99を代入し、Psample=96.27(%)を得た。これは有機相中のアンチピリン含量と等しい。
また、得られた上層(水相)1mLに内標準物質としてトリメチルシリルプロピオン酸ナトリウム−d4(ISOTEC製、10.01mg)を溶解し、水相試料溶液とした。得られた水相試料溶液について表1の条件にて1H−NMRを測定した結果、アンチピリンの特定基のシグナル面積は2.91、トリメチルシリルプロピオン酸ナトリウム−d4の特定基のシグナル面積は240.52であった。なお、アンチピリンの特定基は1,2−ジヒドロ−3H−ピラゾール−3−オンの5位のメチル基、トリメチルシリルプロピオン酸ナトリウム−d4の特定基はメチル基を選択した。
得られた結果に式(3)を適用して水相中のアンチピリン含量(Psample)を算出した。式(3)にIsample=2.91、Hsample=3、Msample=188.23、Wsample=10.90、Istd=240.52、Hstd=9、Mstd=172.24、Wstd=10.01、Pstd=98を代入し、Psample=3.570(%)を得た。これは水相中のアンチピリン含量と等しい。
式(2)に有機相中のアンチピリン含量96.27(%)と水相中のアンチピリン含量3.570(%)を代入し、LogP=1.43を得た。
【0077】
<実施例2〜10>
その他の9化合物についても実施例1と同様にして分配係数を測定した。
【0078】
<実施例11>
NMR法(差し引き法)による、レボフロキサシンの分配係数測定
重水(アクロス製、23mL)とクロロホルム−d(アクロス製、0.03v/v%TMS含有、32mL)を分液漏斗で振り混ぜた後、24時間以上放置し、上層と下層を分離した。得られた上層を飽和重水、下層を飽和クロロホルム−dとした。
レボフロキサシン(東京化成製、21.42mg)に飽和重水2mLを加えて溶解し、さらに飽和クロロホルム−d2mLを加えて30秒間激しく混合後、25℃で24時間振とうし、遠心分離(3000rpm、10分)して上層(水相)と下層(有機相)を完全に分離した。
得られた下層(有機相)1mLに内標準物質として2,6−ジクロロベンズアルデヒド(アクロス製、11.63mg)を溶解し、有機相試料溶液とした。得られた有機相試料溶液について表1の条件にて1H−NMRを測定した結果、レボフロキサシンの特定基のシグナル面積は1.00、2,6−ジクロロベンズアルデヒドの特定基のシグナル面積は2.64であった。なお、レボフロキサシンの特定基はレボフロキサシン5位のプロトン、2,6−ジクロロベンズアルデヒドの特定基はホルミル基を選択した。
得られた結果に式(3)を適用して、有機相中のレボフロキサシン含量(Psample)を算出した。式(3)にIsample=1.00、Hsample=1、Msample=361.37、Wsample=10.71、Istd=2.64、Hstd=1、Mstd=175.01、Wstd=11.63、Pstd=99を代入し、Psample=84.08(%)を得た。これは有機相中のレボフロキサシン含量と等しい。
また、水相中のレボフロキサシン含量は、式(4)により算出した。分配前の含量は、レボフロキサシン10.69mg及び2,6−ジクロロベンズアルデヒド12.39mgを飽和クロロホルム−d1mLに溶解し、表1の条件で1H−NMRを測定及び解析し、式(3)により算出した。1H−NMR測定の結果、レボフロキサシンの特定基のシグナル面積は1.01、2,6−ジクロロベンズアルデヒドの特定基のシグナル面積は2.50であった。式(3)にIsample=1.01、Hsample=1、Msample=361.37、Wsample=10.69、Istd=2.50、Hstd=1、Mstd=175.01、Wstd=12.39、Pstd=99を代入し、分配前のレボフロキサシン含量を算出したところ、Psample=95.72(%)を得た。式(4)に分配前のレボフロキサシン含量=95.72(%)、有機相中のレボフロキサシン含量=84.08(%)を代入し、算出したところ、水相中のレボフロキサシン含量11.64(%)を得た。
式(2)に有機相中のレボフロキサシン含量84.08(%)と水相中のレボフロキサシン含量11.64(%)を代入し、LogP=0.859を得た。
【0079】
<実施例12>
内標準物質を使用しない二重NMR試料管による分配係数測定法(同時測定法)
【0080】
1)補正値の算出
カフェイン(東京化成製、2.29mg)に重水600μL及び重塩酸(アクロス製、20wt.%重水溶液、100μL)を加えて溶解し、カフェイン重水試料溶液とした。 また、カフェイン1.92mgをクロロホルム−d700μLに溶解し、カフェインクロロホルム−d試料溶液とした。
カフェインクロロホルム−d試料溶液を二重NMR試料管(シゲミ製、特殊NMR同軸CELL、5mm)の外管に、カフェイン重水試料溶液を内管に入れ、表1の条件で1H−NMRを測定した。1H−NMR測定の結果、カフェインクロロホルム−d試料溶液の特定基のケミカルシフトは7.508ppm、シグナル面積は8.34であり、カフェイン重水試料溶液の特定基のケミカルシフトは8.806ppm、シグナル面積は99.63であった。なお、カフェインの特定基は1,3,7−トリメチル−3,7−ジヒドロ−プリン−2,6−ジオンの8位のプロトンを選択した。
式(5)にIw’=99.63、Vw=0.7、Ww=2.29、Io’=8.34、Vo=0.7、Wo=1.92を代入し、補正値=10.01を得た。
【0081】
2)分配係数の測定
重水10mLとクロロホルム−d10mLを分液漏斗で振り混ぜた後、24時間以上放置し、上層と下層を分離した。得られた上層を飽和重水、下層を飽和クロロホルム−dとした。
カフェイン21.71mgに飽和クロロホルム−d2mLを加えて溶解し、さらに飽和重水2mLを加えて30秒間激しく混合後、25℃で24時間振とうし、遠心分離(3000rpm、10分)して上層(水相)と下層(有機相)を完全に分離した。得られた上層(水相)600μLに重塩酸100μLを加えて水相試料溶液とし、下層(有機相)はそのまま有機相試料溶液とした。有機相試料溶液を二重NMR試料管の外管に、水相試料溶液を内管に入れ、表1の条件で1H−NMRを測定した。1H−NMR測定の結果、有機相試料溶液中の化合物の特定基のシグナル面積は29.68であり、水相試料溶液中の化合物の特定基のシグナル面積は9.46であった。
式(7)にIo’=29.68、Iw’=9.46、補正値=10.01、希釈率=600/700を代入し、LogP=1.43を得た。
【0082】
従来法(比較例1〜10)及びNMR法(実施例1〜12)の結果を表2及び図1に示す。また、従来法とNMR法のそれぞれで求めた分配係数の相関を図2に示す。
【0083】
表2 分配係数測定結果
【0084】
【表2】
【0085】
NMR法と従来法の分配係数の間には良好な相関が認められた。したがって、NMR法は、分配係数の測定法として信頼に足りる方法といえる。また、NMR法は酸性化合物、中性化合物、塩基性化合物、両性化合物のいずれの化合物にも使用可能であり、さらに、測定可能な分配係数の範囲も広い。なお、NMR測定においてS/N比が大きい化合物ほどNMR法と従来法の分配係数が近くなることが判明した。したがって、有機相又は水相中の化合物濃度が低いためS/N比が小さい化合物については、積算回数を増やすことにより正確な分配係数が得られる。
【0086】
<比較例13>
表3に、NMR法と従来法における所要時間を示す。表中、アンチピリンの従来法は紫外可視吸光光度計、リドカインの従来法はHPLCを用いた測定である。また、分配係数測定に要した時間は、使用前に溶媒を飽和させる操作は含まない。
【0087】
表3 従来法及びNMR法の所要時間
【0088】
【表3】
【0089】
NMR法は従来法と比較して、短時間で分配係数を測定することが可能であり、また、溶媒および化合物の使用量も少ない。
【産業上の利用可能性】
【0090】
本発明により、簡便に分配係数を測定することが可能となった。本発明によれば、測定に使用する化合物は少量で良く、また、迅速に測定することが可能である。また、本発明は、紫外吸収を示さない化合物にも使用可能であるため、適用範囲が広い。
【技術分野】
【0001】
本発明は、NMRによる定量分析工程を含む、分配係数の測定方法に関する。
【背景技術】
【0002】
分配係数は分配比、分配率などともよばれ、化合物の疎水性を表す数値である。分配係数は化合物が水と有機溶媒の二相系に溶解したときの平衡溶解度比であり、下記式(1)
【0003】
【数1】
【0004】
[式中、Pは分配係数、Coは有機相中の化合物の濃度、Cwは水相中の化合物の濃度を表す]
で表される。一般的には、Pの常用対数である下記式(2)
【0005】
【数2】
【0006】
[式中、P、Co、Cwは前述と同義]
が分配係数として広く用いられている。分配係数は化合物の脂溶性を表すため、特に医薬品、農薬及び環境物質の体内吸収や残留性の指標となっている。例えば、化審法では、化合物の物性の指標として1−オクタノールと水の二相系での分配係数が採用されている。また、分配係数の測定法も日本工業規格(非特許文献1)やOECD Test Guideline(非特許文献2)によって標準化されている。
【0007】
ところで、今日用いられている分配係数の測定法(以下、従来法)では、有機相や水相中の化合物濃度を高速液体クロマトグラフィー(HPLC)や紫外可視吸光光度計で定量するため、検量線の作成が不可欠となる。検量線の作成には多量の化合物が必要であるが、化合物が希少である場合や環境への負荷を考慮した場合、化合物の使用量は少ないことが望ましい。また、検量線の作成は作業負荷が大きいため、種々の化合物の分配係数を測定する場合は特に問題となる。さらに従来法は、UVで化合物を定量する測定機器を用いる場合、HPLCや紫外可視吸光光度計で化合物を定量するため、紫外吸収が弱いか、もしくは全くない化合物には使用できないという欠点がある。
【0008】
また、化合物の分配係数を実測するのではなく、コンピューターを用いて分配係数を計算する方法も報告されているが、計算値は実測値と乖離することがあるため、信頼性の面から実測することが好ましい。
【0009】
ところで、近年、NMRを用いた化合物の定量分析法が報告されている(非特許文献3)。NMRを用いた定量分析法のひとつである内部標準法では、化合物と内標準物質の両方を含有する溶液をNMRで測定し、NMRスペクトル上の化合物と内標準物質のそれぞれのシグナル面積を比較することで、化合物の定量を可能としている。
【先行技術文献】
【非特許文献】
【0010】
【非特許文献1】日本工業規格Z7260−107(2000)、「分配係数(1−オクタノール/水)の測定−フラスコ振とう法」.
【非特許文献2】OECD Test Guideline、(OECD理事会決定「C(81)30最終別添1」)107.
【非特許文献3】平成21年度成果報告書 基準認証研究開発事業「1対多型校正技術の研究開発」 平成22年3月経済産業省産業技術環境局知的基盤課 委託先(独)産業技術総合研究所
【発明の概要】
【発明が解決しようとする課題】
【0011】
従来の分配係数の測定法は、水相中及び有機相中の化合物の定量のために検量線の作成が必要である。しかしながら、検量線の作成には多量の化合物が必要であるため、環境への負荷が大きい。また、作業負荷が大きいことから簡便性、迅速性に欠ける点も課題である。また、UV検出器を備えたHPLC又は紫外可視吸光光度計で測定する場合、紫外吸収が弱い若しくは全くない化合物には使用できない点も課題である。
【課題を解決するための手段】
【0012】
本発明は、水相中及び有機相中の化合物の定量にNMRを用いることで上記課題を解決した(以下、NMR法)。すなわち、本発明は以下の通りである。
【0013】
第1発明は、NMRを用いた定量分析工程を含む、分配係数の測定方法に関する。
第2発明は、以下の工程を含む分配係数の測定方法であって、
i)化合物、重水及び重水素化有機溶媒を混合する工程、
ii)工程iの混合物を水相及び有機相に分離する工程、及び
iii)工程iiの水相及び/又は有機相中の化合物の特定基のシグナル面積をNMRを用いて測定する工程、
を含む、第1発明記載の分配係数の測定方法に関する。
第3発明は、工程iiiが、水相及び有機相中の化合物の特定基のシグナル面積をNMRを用いて測定する工程である、第2発明記載の分配係数の測定方法に関する。
【0014】
第4発明は、以下の工程を含む分配係数の測定方法であって、
i)化合物、飽和重水及び飽和重水素化有機溶媒を混合する工程、
ii)工程iの混合物を水相及び有機相に分離する工程、
iii)工程iiで得た有機相に内標準物質を溶解させ有機相試料溶液とした後、NMRを用いて有機相試料溶液中の化合物及び内標準物質の特定基のシグナル面積を測定し、当該シグナル面積を用いて有機相中の化合物含量を算出する工程、
iv)工程iiで得た水相に工程iiiと同一又は異なる内標準物質を溶解させ水相試料溶液とした後、NMRを用いて水相試料溶液中の化合物及び内標準物質の特定基のシグナル面積を測定し、当該シグナル面積を用いて水相中の化合物含量を算出する工程、及び
v)工程iiiで算出した有機相中の化合物含量と、工程ivで算出した水相中の化合物含量を用いて化合物の分配係数を算出する工程、
を含む、第3発明記載の分配係数の測定方法に関する。
【0015】
第5発明は、以下の工程を含む分配係数の測定方法であって、
i)化合物、飽和重水及び飽和重水素化有機溶媒を混合する工程、
ii)工程iの混合物を水相及び有機相に分離する工程、
iii)工程iiで得た水相に重水素化酸、重水素化塩基若しくはシフト試薬を溶解させ水相試料溶液とし、工程iiで得た有機相を有機相試料溶液とするか、或いは工程iiで得た水相を水相試料溶液とし、工程iiで得た有機相にシフト試薬を溶解させ有機相試料溶液とし、当該有機相試料溶液と水相試料溶液のうち一方を二重NMR管の外管に採取し、他方を二重NMR管の内管に採取した後、NMRを用いて有機相試料溶液及び水相試料溶液中の化合物の特定基のシグナル面積を測定する工程、及び
iv)工程iiiで測定した有機相試料溶液及び水相試料溶液中の化合物の特定基のシグナル面積を用いて化合物の分配係数を算出する工程、
を含む、第3発明記載の分配係数の測定方法に関する。
【発明の効果】
【0016】
本発明によれば、検量線の作成が不要であること等から、従来法と比較して少量の化合物で分配係数を測定することが可能である。また、簡便、迅速に分配係数を測定することが可能である。さらに、本発明によれば、紫外吸収を示さない化合物の分配係数の測定も可能である。
【図面の簡単な説明】
【0017】
【図1】横軸に化合物の種類、縦軸に化合物の分配係数をとり、従来法とNMR法で測定した各種化合物の分配係数を示したグラフである。
【図2】横軸に分配係数(従来法)、縦軸に分配係数(NMR法)をとり、従来法とNMR法で測定した分配係数の相関性を示したグラフである。
【発明を実施するための形態】
【0018】
本明細書中において使用される語句の定義は以下の通りである。
【0019】
化合物とは、酸性化合物、中性化合物、塩基性化合物、両性化合物等の任意の化合物を表す。なお、本発明においては、コンピューター(Pallas3.0、インフォコム株式会社)を用いて計算し、酸塩基解離定数が6以下の化合物を酸性化合物、7.8以上の化合物を塩基性化合物、両方の性質を持ち合わせるものを両性化合物とし、それ以外を中性化合物と分類した。
【0020】
重水とは、水分子の二つの水素原子を重水素原子に置き換えた水を表し、D2Oと表すこともある。
【0021】
重水素化有機溶媒とは、有機溶媒分子に含まれる水素原子の一部または全部を重水素原子に置き換えた有機溶媒を表す。重水素化有機溶媒としては、例えば、シクロヘキサン−d12、トルエン−d8、ベンゼン−d6、クロロホルム−d、ジクロロメタン−d2又は1−オクタノール−d18が挙げられる。
【0022】
内標準物質とは、化合物を定量するために、水相及び/又は有機相に添加する物質である。内標準物質としては、例えば、2,6−ジクロロベンズアルデヒド、2−ヒドロキシ−3,5−ジニトロ−安息香酸、テレフタル酸ジメチルエステル、フタル酸水素カリウム、安息香酸、1,3,5−トリオキサン、テトラメチルピラジン、ジメチルスルホン、酢酸ナトリウム、トリメチルシリルプロパンスルホン酸ナトリウム−d6、トリメチルシリルプロピオン酸ナトリウム−d4又は1,4−ビス(トリメチルシリル)ベンゼン−d4が挙げられる。
【0023】
シグナル面積は、ピーク面積又は積分値と同義である。
【0024】
特定基とは、化合物および内標準物質のそれぞれの任意の置換基を表す。
【0025】
飽和重水素化有機溶媒とは、重水が飽和した重水素化有機溶媒を表す。
【0026】
飽和重水とは、重水素化有機溶媒が飽和した重水を表す。
【0027】
重水素化酸とは、酸分子に含まれる水素原子の一部または全部を重水素原子に置き換えた酸を表す。重水素化酸としては、例えば、蟻酸−d2、酢酸−d、酢酸−d4、トリフルオロ酢酸−d、塩酸−d(重塩酸)、硫酸−d2が挙げられる。
【0028】
重水素化塩基とは、塩基分子に含まれる水素原子の一部または全部を重水素原子に置き換えた塩基を表す。重水素化塩基としては、例えば、水酸化アンモニウム−d5、ピリジン−d5、水酸化ナトリウム−d、水酸化カリウム−dが挙げられる。
【0029】
シフト試薬とは、有機配位子に配位結合した希土(ランタニド)類系列のイオンである。例えば、トリス(2、2、6、6−テトラメチル−3、5−ヘプタンジオナト)ユウロピウム (Eu(dpm)3)、トリス(6、6、7、7、8、8、8−ヘプタフルオロ−2、2−ジメチル−3、5−オクタンジオナト)ユウロピウム (Eu(fod)3)、トリス[N、N−ビス(トリメチルシリル)アミド]ユウロピウム、トリス(4、4、4−トリフルオロ−1−(2−チエニル)−1、3−ブタンジオノ)ユウロピウム、トリス(6、6、7、7、8、8、8−ヘプタフルオロ−2、2−ジメチル−3、5−オクタンジオナト)イッテルビウム (Yb(fod)3)、トリス(2、2、6、6−テトラメチル−3、5−ヘプタンジオナト)ジスプロシウム(III) (Dy(dpm)3)、トリス(2、2、6、6‐テトラメチル‐3、5‐ヘプタンジオナト)ガドリニウム (Gd(dpm)3)が挙げられる。
【0030】
二重NMR試料管とは、外管(アウターチューブ)と内管(インナーチューブ)を備えたNMR管を表す。外管と内管の各々に試料溶液を採取することができる。
【0031】
本発明の好ましい形態は以下の通りである。
【0032】
化合物は、中性化合物及び両性化合物が、濃度による分配係数への影響が少ないため好ましい。また、より正確な分配係数が得られるため、LogPが−3〜3の範囲の化合物が好ましく、−2.5〜2.5の範囲の化合物が好ましく、−2〜2の範囲の化合物が特に好ましい。
【0033】
化合物の濃度は、重水素化有機溶媒1mLに対し、好ましくは0.008〜30 mgであり、更に好ましくは0.02〜10 mgであり、特に好ましくは1.8〜10 mgである。また、重水1mLに対し、好ましくは0.008〜30mgであり、更に好ましくは0.02〜10mgであり、特に好ましくは1.1〜10mgである。
【0034】
NMRは、測定感度の点からプロトンNMR(1H−NMR)が好ましい。
【0035】
重水素化有機溶媒は、有機溶媒分子に含まれる水素原子の全部を重水素原子に置き換えた有機溶媒が好ましい。好ましくはシクロヘキサン−d12、トルエン−d8、ベンゼン−d6、クロロホルム−d、ジクロロメタン−d2又は1−オクタノール−d18であり、更に好ましくはクロロホルム−d又は1−オクタノール−d18であり、特に好ましくはクロロホルム−dである。
【0036】
内標準物質は、そのNMRシグナルのうち少なくとも1つのシグナルが、化合物のNMRシグナルと重ならないものであれば、任意に選ぶことができる。有機相に添加する場合、好ましくは2,6−ジクロロベンズアルデヒド又は1,4−ビス(トリメチルシリル)ベンゼン−d4である。水相に添加する場合、好ましくはトリメチルシリルプロピオン酸ナトリウム-d4又はトリメチルシリルプロパンスルホン酸ナトリウム−d6である。
【0037】
重水素化酸は、好ましくは蟻酸−d2、酢酸−d、酢酸−d4、トリフルオロ酢酸−d、塩酸−d(重塩酸)、硫酸−d2であり、更に好ましくは酢酸−d4、トリフルオロ酢酸−d、塩酸−d(重塩酸)であり、特に好ましくは塩酸−d(重塩酸)である。
【0038】
重水素化塩基は、好ましくは水酸化アンモニウム−d5、ピリジン−d5、水酸化ナトリウム−d、水酸化カリウム−dである。
【0039】
シフト試薬は、好ましくはトリス(2、2、6、6−テトラメチル−3、5−ヘプタンジオナト)ユウロピウム、トリス(6、6、7、7、8、8、8−ヘプタフルオロ−2、2−ジメチル−3、5−オクタンジオナト)ユウロピウムである。
【0040】
特定基は任意に選択できるが、精確なシグナル面積を得るため、そのNMRシグナルが他のNMRシグナルと完全に分離している置換基から選ぶことが好ましい。
【0041】
以下に本発明の一般的な実施形態を説明するが、本実施形態によって本発明の範囲が限定されるものではない。
【0042】
(工程1)飽和溶媒の作成
適当量の重水素化有機溶媒と重水を激しく振り混ぜた後、室温下、24時間以上静置する。その後、二つの層を分離し、飽和重水素化有機溶媒と飽和重水を得る。
【0043】
(工程2)化合物の分配操作
化合物を精秤し、これに工程1で得られた飽和重水素化有機溶媒を一定量正確に加えて化合物を完全に溶解させる。これに工程1で得られた飽和重水を、飽和重水素化有機溶媒と同容量加え、激しく混合後、25℃で24時間振とうする。これを遠心分離により二つの層に分離し、有機相と水相を得る。
【0044】
(工程3)NMRによる、有機相及び/又は水相中の化合物の定量分析と分配係数の算出
有機相及び水相中の化合物の定量分析は、以下に示す個別定量法又は同時定量法により行うことができる。
【0045】
1.個別定量法
本方法では、工程2で得られた有機相及び/又は水相に内標準物質を添加し、得られた有機相試料溶液及び水相試料溶液についてNMRを用いて個別に定量分析を行った後、分配係数を算出する。
【0046】
1)有機相中の化合物の定量分析
内標準物質を精秤し、これに工程2で得られた有機相を一定量正確に加えて内標準物質を完全に溶解させ、これを有機相試料溶液とする。その後、有機相試料溶液をNMRで測定する。この際、公知技術(平成21年度成果報告書 基準認証研究開発事業「1対多型校正技術の研究開発」 平成22年3月経済産業省産業技術環境局知的基盤課 委託先(独)産業技術総合研究所)等を参考にして測定条件を選択し、化合物及び内標準物質の特定基のシグナル面積を得る。下記式(3)
【0047】
【数3】
【0048】
に化合物及び内標準物質の特定基のシグナル面積、特定基のプロトン数、分子量及びNMR測定に用いた質量並びに内標準物質の純度を代入することにより、有機相中の化合物の含量を算出する。
【0049】
2)水相中の化合物の定量分析
工程2で得られた水相中の化合物の定量分析は、以下に示す実測法又は差し引き法により行うことができる。適用可能な分配係数の範囲が広いこと及び正確な分配係数を得られることから実測法が好ましい。
<実測法>
工程2で得られた水相と、内標準物質を用いて、1)と同様の方法により水相試料溶液を作成後、NMRで測定し、式(3)に従って水相中の化合物含量を算出する。
<差し引き法>
下記式(4)
【0050】
【数4】
【0051】
により水相中の化合物含量を算出することができる。ここで、分配前の化合物含量とは、工程2の分配前の化合物純度を表す。例えば、工程2と同量の化合物と、一定量の内標準物質を精量し、これに工程2と同量の飽和重水素化有機溶媒を加えて完全に溶解させ試料溶液とした後、1)と同様の方法によりNMRを測定し、式(3)に従って算出する。このようにして得た分配前の化合物含量と、1)で得た有機相中の化合物含量を式(4)に代入し、水相中の化合物含量を算出する。
【0052】
3)分配係数の算出
化合物含量の比は化合物濃度の比と等しいため、式(2)のCoに1)で算出した有機相中の化合物含量を代入し、Cwに2)で算出した水相中の化合物含量を代入することで、化合物の分配係数を算出する。
【0053】
2.同時定量法
本方法では、工程2で得られた有機相又は水相に、重水素化酸、重水素化塩基若しくはシフト試薬を加え、得られた有機相試料溶液及び水相試料溶液の一方を二重NMR試料管の外管に、他方を二重NMR試料管の内管に採取し、NMRを用いて定量分析を行った後、分配係数を算出する。本測定法は内標準物質を必要とせず、また、有機相試料溶液と水相試料溶液を同時に測定できるため、簡便である。
【0054】
1)補正値の算出
同一の化合物濃度を有する有機相試料溶液と水相試料溶液であっても、一方を二重NMR管の外管に、他方を二重NMR管の内管に採取し、NMR測定した場合、種々の要因により化合物の特定基のシグナル面積に差異が生じる。したがって、当該差異を補正するための補正値を算出する。
化合物を重水素化有機溶媒に溶解し有機相試料溶液とする。また、有機相試料溶液中の化合物濃度と等しくなるように化合物及び重水素化酸、重水素化塩基若しくはシフト試薬を重水に溶解し水相試料溶液とする。或いは化合物及びシフト試薬を重水素化有機溶媒に溶解し有機相試料溶液とする。また、有機相試料溶液中の化合物濃度と等しくなるように化合物を重水に溶解し水相試料溶液とする。
有機相試料溶液を二重NMR試料管の外管に、水相試料溶液を二重NMR試料管の内管に採取し、NMRを測定し、水相試料溶液及び有機相試料溶液中の化合物の特定基のシグナル面積を得る。溶媒量、化合物の質量及びシグナル面積を下記式(5)
【0055】
【数5】
【0056】
に代入し、補正値を算出する。
【0057】
2)化合物の定量分析
工程2で得られた水相に重水素化酸、重水素化塩基若しくはシフト試薬を加えて水相試料溶液とし、工程2で得られた有機相はそのまま有機相試料溶液とする。或いは工程2で得られた水相はそのまま水相試料溶液とし、工程2で得られた有機相にシフト試薬を加えて有機相試料溶液とする。有機相試料溶液を二重NMR試料管の外管に、水相試料溶液を二重NMR試料管の内管に入れ、NMRを測定し、水相試料溶液及び有機相試料溶液中の化合物の特定基のシグナル面積を得る。
【0058】
3)分配係数の算出
化合物の特定基のシグナル面積は化合物濃度に比例する。したがって、シグナル面積の比は化合物濃度の比と等しい。すなわち、分配係数は下記式(6)
【0059】
【数6】
【0060】
により算出できる。
ここで、2)において水相に加える重水素化酸、重水素化塩基若しくはシフト試薬が水溶液の場合、水相は当該水溶液により希釈されていることから、水相試料溶液中の化合物の特定基のシグナル面積は、水相中の化合物の特定基のシグナル面積よりも希釈率分だけ小さい値となる。また、1)で述べたように、有機相試料溶液中の化合物の特定基のシグナル面積は補正する必要がある。これらを考慮し、分配係数は下記式(7)
【0061】
【数7】
【0062】
[式中、P、Io’、Iw’は前述と同義]
により算出できる。式(7)に1)で得られた補正値、2)で得られた化合物の特定基のシグナル面積、及び希釈率を代入することで、化合物の分配係数を算出する。なお、水相に加える重水素化酸、重水素化塩基若しくはシフト試薬が水溶液ではない場合は、希釈率は考慮しなくてよい。
【0063】
以下に本発明を実施例及び比較例にて更に詳細に説明するが、これらによって本発明の範囲が限定されるものではない。
【実施例】
【0064】
<比較例1>
従来法による、アンチピリンの分配係数測定
【0065】
1.検量線の作成
以下、クロロホルム溶液及び水溶液中のアンチピリン濃度に関する検量線を作成した。
1)クロロホルム溶液中のアンチピリン濃度に関する検量線
アンチピリン(シグマ・アルドリッチ製、10.67mg)にクロロホルム(関東化学製、特級)を加えて25mLとし、原液(426.8μg/mL)とした。原液2mLにクロロホルムを加えて20mLとし、溶液Aとした(42.68μg/mL)。溶液A4mLにクロロホルムを加えて20mLとし、溶液Bとした(8.536μg/mL)。溶液B4mLにクロロホルムを加えて20mLとし、溶液Cとした(1.707μg/mL)。溶液A7mLにクロロホルムを加えて10mLとし、溶液Dとした(29.88μg/mL)。溶液B4mLにクロロホルムを加えて10mLとし、溶液Eとした(3.414μg/mL)。溶液C4mLにクロロホルムを加えて10mLとし、溶液Fとした(0.6829μg/mL)。
溶液AからFを紫外可視吸光光度計で測定し、約278nmにおける吸光度について、x軸を濃度、y軸を吸光度とした回帰式を作成し、相関係数1.000、切片0.001564、傾き0.05176の下記式(8)
【0066】
【数8】
【0067】
を得た。
【0068】
2)水溶液中のアンチピリン濃度に関する検量線
アンチピリン(シグマ・アルドリッチ製、20.44mg)に超純水(ミリQ水)を加えて50mLとし、原液(408.8μg/mL)とした。原液2mLに超純水を加えて20mLとし、溶液Aとした(40.88μg/mL)。溶液A4mLに超純水を加えて20mLとし、溶液Bとした(8.176μg/mL)。溶液B4mLに超純水を加えて20mLとし、溶液Cとした(1.635μg/mL)。溶液A5mLに超純水を加えて10mLとし、溶液Dとした(20.44μg/mL)。溶液B4mLに超純水を加えて10mLとし、溶液Eとした(3.270μg/mL)。溶液C4mLに超純水を加えて10mLとし、溶液Fとした(0.6541μg/mL)。
溶液AからFを紫外可視吸光光度計で測定し、約242nmにおける吸光度について、x軸を濃度、y軸を吸光度とした回帰式を作成し、相関係数1.000、切片0.001521、傾き0.04860の下記式(9)
【0069】
【数9】
【0070】
を得た。
【0071】
2.分配係数測定
以下、クロロホルム/水系での分配係数を従来法により測定した。
超純水(ミリQ水、200mL)とクロロホルム(関東化学製、特級、200mL)を分液漏斗で振り混ぜた後、24時間以上放置し、上層と下層を分離した。得られた上層を飽和水、下層を飽和クロロホルムとした。
アンチピリン(シグマ・アルドリッチ製、201.27mg)に飽和クロロホルムを加えて20mLとし,アンチピリン溶液とした。アンチピリン溶液12mLに飽和水12mLを加えて室温で30分間激しく振とう後、25℃で24時間振とうし、遠心分離(3000rpm、10分)して上層(水相)と下層(有機相)を完全に分離した。得られた上層(水相)4mLに超純水を加えて20mLとし、溶液Aとした。溶液A 4mLに超純水を加えて20mLとし、水相試料溶液とした。また、得られた下層(有機相)2mLにクロロホルムを加えて10mLとし、溶液Bとした。溶液B 2mLにクロロホルムを加えて20mLとし、溶液Cとした。溶液C 2mLにクロロホルムを加えて20mLとし、有機相試料溶液とした。
有機相試料溶液と水相試料溶液の吸光度を紫外可視吸光光度計で測定した(有機相試料溶液:278nm、水相試料溶液:242nm)。それぞれの試料溶液から得られた吸光度を式(8)及び式(9)に代入し、有機相試料溶液中のアンチピリン濃度(Co)と水相試料溶液中のアンチピリン濃度(Cw)を定量した。得られたCo及びCwを式(2)に代入し、LogP=1.35を得た。
【0072】
<比較例2〜10>
その他9化合物についても比較例1と同様にして分配係数を測定した。
【0073】
<実施例1>
NMR法(実測法)による、アンチピリンの分配係数測定
重水(アクロス製、17mL)とクロロホルム−d(アクロス製、0.03v/v%TMS含有、17mL)を分液漏斗で振り混ぜた後、24時間以上放置し、上層と下層を分離した。得られた上層を飽和重水、下層を飽和クロロホルム−dとした。
アンチピリン(シグマ・アルドリッチ製、21.79mg)に飽和クロロホルム−d2mLを加えて溶解し、さらに飽和重水2mLを加えて30秒間激しく混合後、25℃で24時間振とうし、遠心分離(3000rpm、10分)して上層(水相)と下層(有機相)を完全に分離した。
得られた下層(有機相)1mLに内標準物質として2,6−ジクロロベンズアルデヒド(アクロス製、10.84mg)を溶解し、有機相試料溶液とした。得られた有機相試料溶液について表1の条件にて1H−NMRを測定した結果、アンチピリンの特定基のシグナル面積は3.00、2,6−ジクロロベンズアルデヒドの特定基のシグナル面積は1.10であった。なお、アンチピリンの特定基は1,2−ジヒドロ−3H−ピラゾール−3−オンの5位のメチル基、2,6−ジクロロベンズアルデヒドの特定基はホルミル基を選択した。
【0074】
表1 1H−NMR測定条件及び解析条件
【0075】
【表1】
【0076】
得られた結果に式(3)を適用して、有機相中のアンチピリン含量(Psample)を算出した。式(3)にIsample=3.00、Hsample=3、Msample=188.23、Wsample=10.90、Istd=1.10、Hstd=1、Mstd=175.01、Wstd=10.84、Pstd=99を代入し、Psample=96.27(%)を得た。これは有機相中のアンチピリン含量と等しい。
また、得られた上層(水相)1mLに内標準物質としてトリメチルシリルプロピオン酸ナトリウム−d4(ISOTEC製、10.01mg)を溶解し、水相試料溶液とした。得られた水相試料溶液について表1の条件にて1H−NMRを測定した結果、アンチピリンの特定基のシグナル面積は2.91、トリメチルシリルプロピオン酸ナトリウム−d4の特定基のシグナル面積は240.52であった。なお、アンチピリンの特定基は1,2−ジヒドロ−3H−ピラゾール−3−オンの5位のメチル基、トリメチルシリルプロピオン酸ナトリウム−d4の特定基はメチル基を選択した。
得られた結果に式(3)を適用して水相中のアンチピリン含量(Psample)を算出した。式(3)にIsample=2.91、Hsample=3、Msample=188.23、Wsample=10.90、Istd=240.52、Hstd=9、Mstd=172.24、Wstd=10.01、Pstd=98を代入し、Psample=3.570(%)を得た。これは水相中のアンチピリン含量と等しい。
式(2)に有機相中のアンチピリン含量96.27(%)と水相中のアンチピリン含量3.570(%)を代入し、LogP=1.43を得た。
【0077】
<実施例2〜10>
その他の9化合物についても実施例1と同様にして分配係数を測定した。
【0078】
<実施例11>
NMR法(差し引き法)による、レボフロキサシンの分配係数測定
重水(アクロス製、23mL)とクロロホルム−d(アクロス製、0.03v/v%TMS含有、32mL)を分液漏斗で振り混ぜた後、24時間以上放置し、上層と下層を分離した。得られた上層を飽和重水、下層を飽和クロロホルム−dとした。
レボフロキサシン(東京化成製、21.42mg)に飽和重水2mLを加えて溶解し、さらに飽和クロロホルム−d2mLを加えて30秒間激しく混合後、25℃で24時間振とうし、遠心分離(3000rpm、10分)して上層(水相)と下層(有機相)を完全に分離した。
得られた下層(有機相)1mLに内標準物質として2,6−ジクロロベンズアルデヒド(アクロス製、11.63mg)を溶解し、有機相試料溶液とした。得られた有機相試料溶液について表1の条件にて1H−NMRを測定した結果、レボフロキサシンの特定基のシグナル面積は1.00、2,6−ジクロロベンズアルデヒドの特定基のシグナル面積は2.64であった。なお、レボフロキサシンの特定基はレボフロキサシン5位のプロトン、2,6−ジクロロベンズアルデヒドの特定基はホルミル基を選択した。
得られた結果に式(3)を適用して、有機相中のレボフロキサシン含量(Psample)を算出した。式(3)にIsample=1.00、Hsample=1、Msample=361.37、Wsample=10.71、Istd=2.64、Hstd=1、Mstd=175.01、Wstd=11.63、Pstd=99を代入し、Psample=84.08(%)を得た。これは有機相中のレボフロキサシン含量と等しい。
また、水相中のレボフロキサシン含量は、式(4)により算出した。分配前の含量は、レボフロキサシン10.69mg及び2,6−ジクロロベンズアルデヒド12.39mgを飽和クロロホルム−d1mLに溶解し、表1の条件で1H−NMRを測定及び解析し、式(3)により算出した。1H−NMR測定の結果、レボフロキサシンの特定基のシグナル面積は1.01、2,6−ジクロロベンズアルデヒドの特定基のシグナル面積は2.50であった。式(3)にIsample=1.01、Hsample=1、Msample=361.37、Wsample=10.69、Istd=2.50、Hstd=1、Mstd=175.01、Wstd=12.39、Pstd=99を代入し、分配前のレボフロキサシン含量を算出したところ、Psample=95.72(%)を得た。式(4)に分配前のレボフロキサシン含量=95.72(%)、有機相中のレボフロキサシン含量=84.08(%)を代入し、算出したところ、水相中のレボフロキサシン含量11.64(%)を得た。
式(2)に有機相中のレボフロキサシン含量84.08(%)と水相中のレボフロキサシン含量11.64(%)を代入し、LogP=0.859を得た。
【0079】
<実施例12>
内標準物質を使用しない二重NMR試料管による分配係数測定法(同時測定法)
【0080】
1)補正値の算出
カフェイン(東京化成製、2.29mg)に重水600μL及び重塩酸(アクロス製、20wt.%重水溶液、100μL)を加えて溶解し、カフェイン重水試料溶液とした。 また、カフェイン1.92mgをクロロホルム−d700μLに溶解し、カフェインクロロホルム−d試料溶液とした。
カフェインクロロホルム−d試料溶液を二重NMR試料管(シゲミ製、特殊NMR同軸CELL、5mm)の外管に、カフェイン重水試料溶液を内管に入れ、表1の条件で1H−NMRを測定した。1H−NMR測定の結果、カフェインクロロホルム−d試料溶液の特定基のケミカルシフトは7.508ppm、シグナル面積は8.34であり、カフェイン重水試料溶液の特定基のケミカルシフトは8.806ppm、シグナル面積は99.63であった。なお、カフェインの特定基は1,3,7−トリメチル−3,7−ジヒドロ−プリン−2,6−ジオンの8位のプロトンを選択した。
式(5)にIw’=99.63、Vw=0.7、Ww=2.29、Io’=8.34、Vo=0.7、Wo=1.92を代入し、補正値=10.01を得た。
【0081】
2)分配係数の測定
重水10mLとクロロホルム−d10mLを分液漏斗で振り混ぜた後、24時間以上放置し、上層と下層を分離した。得られた上層を飽和重水、下層を飽和クロロホルム−dとした。
カフェイン21.71mgに飽和クロロホルム−d2mLを加えて溶解し、さらに飽和重水2mLを加えて30秒間激しく混合後、25℃で24時間振とうし、遠心分離(3000rpm、10分)して上層(水相)と下層(有機相)を完全に分離した。得られた上層(水相)600μLに重塩酸100μLを加えて水相試料溶液とし、下層(有機相)はそのまま有機相試料溶液とした。有機相試料溶液を二重NMR試料管の外管に、水相試料溶液を内管に入れ、表1の条件で1H−NMRを測定した。1H−NMR測定の結果、有機相試料溶液中の化合物の特定基のシグナル面積は29.68であり、水相試料溶液中の化合物の特定基のシグナル面積は9.46であった。
式(7)にIo’=29.68、Iw’=9.46、補正値=10.01、希釈率=600/700を代入し、LogP=1.43を得た。
【0082】
従来法(比較例1〜10)及びNMR法(実施例1〜12)の結果を表2及び図1に示す。また、従来法とNMR法のそれぞれで求めた分配係数の相関を図2に示す。
【0083】
表2 分配係数測定結果
【0084】
【表2】
【0085】
NMR法と従来法の分配係数の間には良好な相関が認められた。したがって、NMR法は、分配係数の測定法として信頼に足りる方法といえる。また、NMR法は酸性化合物、中性化合物、塩基性化合物、両性化合物のいずれの化合物にも使用可能であり、さらに、測定可能な分配係数の範囲も広い。なお、NMR測定においてS/N比が大きい化合物ほどNMR法と従来法の分配係数が近くなることが判明した。したがって、有機相又は水相中の化合物濃度が低いためS/N比が小さい化合物については、積算回数を増やすことにより正確な分配係数が得られる。
【0086】
<比較例13>
表3に、NMR法と従来法における所要時間を示す。表中、アンチピリンの従来法は紫外可視吸光光度計、リドカインの従来法はHPLCを用いた測定である。また、分配係数測定に要した時間は、使用前に溶媒を飽和させる操作は含まない。
【0087】
表3 従来法及びNMR法の所要時間
【0088】
【表3】
【0089】
NMR法は従来法と比較して、短時間で分配係数を測定することが可能であり、また、溶媒および化合物の使用量も少ない。
【産業上の利用可能性】
【0090】
本発明により、簡便に分配係数を測定することが可能となった。本発明によれば、測定に使用する化合物は少量で良く、また、迅速に測定することが可能である。また、本発明は、紫外吸収を示さない化合物にも使用可能であるため、適用範囲が広い。
【特許請求の範囲】
【請求項1】
NMRを用いた定量分析工程を含む、分配係数の測定方法。
【請求項2】
以下の工程を含む分配係数の測定方法であって、
i)化合物、重水及び重水素化有機溶媒を混合する工程、
ii)工程iの混合物を水相及び有機相に分離する工程、及び
iii)工程iiの水相及び/又は有機相中の化合物の特定基のシグナル面積をNMRを用いて測定する工程、
を含む、請求項1記載の分配係数の測定方法。
【請求項3】
工程iiiが、水相及び有機相中の化合物の特定基のシグナル面積をNMRを用いて測定する工程である、請求項2記載の分配係数の測定方法。
【請求項4】
以下の工程を含む分配係数の測定方法であって、
i)化合物、飽和重水及び飽和重水素化有機溶媒を混合する工程、
ii)工程iの混合物を水相及び有機相に分離する工程、
iii)工程iiで得た有機相に内標準物質を溶解させ有機相試料溶液とした後、NMRを用いて有機相試料溶液中の化合物及び内標準物質の特定基のシグナル面積を測定し、当該シグナル面積を用いて有機相中の化合物含量を算出する工程、
iv)工程iiで得た水相に工程iiiと同一又は異なる内標準物質を溶解させ水相試料溶液とした後、NMRを用いて水相試料溶液中の化合物及び内標準物質の特定基のシグナル面積を測定し、当該シグナル面積を用いて水相中の化合物含量を算出する工程、及び
v)工程iiiで算出した有機相中の化合物含量と、工程ivで算出した水相中の化合物含量を用いて化合物の分配係数を算出する工程、
を含む、請求項3記載の分配係数の測定方法。
【請求項5】
以下の工程を含む分配係数の測定方法であって、
i)化合物、飽和重水及び飽和重水素化有機溶媒を混合する工程、
ii)工程iの混合物を水相及び有機相に分離する工程、
iii)工程iiで得た水相に重水素化酸、重水素化塩基若しくはシフト試薬を溶解させ水相試料溶液とし、工程iiで得た有機相を有機相試料溶液とするか、或いは工程iiで得た水相を水相試料溶液とし、工程iiで得た有機相にシフト試薬を溶解させ有機相試料溶液とし、当該有機相試料溶液と水相試料溶液のうち一方を二重NMR管の外管に採取し、他方を二重NMR管の内管に採取した後、NMRを用いて有機相試料溶液及び水相試料溶液中の化合物の特定基のシグナル面積を測定する工程、及び
iv)工程iiiで測定した有機相試料溶液及び水相試料溶液中の化合物の特定基のシグナル面積を用いて化合物の分配係数を算出する工程、
を含む、請求項3記載の分配係数の測定方法。
【請求項1】
NMRを用いた定量分析工程を含む、分配係数の測定方法。
【請求項2】
以下の工程を含む分配係数の測定方法であって、
i)化合物、重水及び重水素化有機溶媒を混合する工程、
ii)工程iの混合物を水相及び有機相に分離する工程、及び
iii)工程iiの水相及び/又は有機相中の化合物の特定基のシグナル面積をNMRを用いて測定する工程、
を含む、請求項1記載の分配係数の測定方法。
【請求項3】
工程iiiが、水相及び有機相中の化合物の特定基のシグナル面積をNMRを用いて測定する工程である、請求項2記載の分配係数の測定方法。
【請求項4】
以下の工程を含む分配係数の測定方法であって、
i)化合物、飽和重水及び飽和重水素化有機溶媒を混合する工程、
ii)工程iの混合物を水相及び有機相に分離する工程、
iii)工程iiで得た有機相に内標準物質を溶解させ有機相試料溶液とした後、NMRを用いて有機相試料溶液中の化合物及び内標準物質の特定基のシグナル面積を測定し、当該シグナル面積を用いて有機相中の化合物含量を算出する工程、
iv)工程iiで得た水相に工程iiiと同一又は異なる内標準物質を溶解させ水相試料溶液とした後、NMRを用いて水相試料溶液中の化合物及び内標準物質の特定基のシグナル面積を測定し、当該シグナル面積を用いて水相中の化合物含量を算出する工程、及び
v)工程iiiで算出した有機相中の化合物含量と、工程ivで算出した水相中の化合物含量を用いて化合物の分配係数を算出する工程、
を含む、請求項3記載の分配係数の測定方法。
【請求項5】
以下の工程を含む分配係数の測定方法であって、
i)化合物、飽和重水及び飽和重水素化有機溶媒を混合する工程、
ii)工程iの混合物を水相及び有機相に分離する工程、
iii)工程iiで得た水相に重水素化酸、重水素化塩基若しくはシフト試薬を溶解させ水相試料溶液とし、工程iiで得た有機相を有機相試料溶液とするか、或いは工程iiで得た水相を水相試料溶液とし、工程iiで得た有機相にシフト試薬を溶解させ有機相試料溶液とし、当該有機相試料溶液と水相試料溶液のうち一方を二重NMR管の外管に採取し、他方を二重NMR管の内管に採取した後、NMRを用いて有機相試料溶液及び水相試料溶液中の化合物の特定基のシグナル面積を測定する工程、及び
iv)工程iiiで測定した有機相試料溶液及び水相試料溶液中の化合物の特定基のシグナル面積を用いて化合物の分配係数を算出する工程、
を含む、請求項3記載の分配係数の測定方法。
【図1】

【図2】
【図2】
【公開番号】特開2012−177643(P2012−177643A)
【公開日】平成24年9月13日(2012.9.13)
【国際特許分類】
【出願番号】特願2011−41393(P2011−41393)
【出願日】平成23年2月28日(2011.2.28)
【出願人】(000001395)杏林製薬株式会社 (120)
【公開日】平成24年9月13日(2012.9.13)
【国際特許分類】
【出願日】平成23年2月28日(2011.2.28)
【出願人】(000001395)杏林製薬株式会社 (120)
[ Back to top ]